

Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones
- PMID: 20410102
- PMCID: PMC3278277
- DOI: 10.1523/JNEUROSCI.6383-09.2010
Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones
Abstract
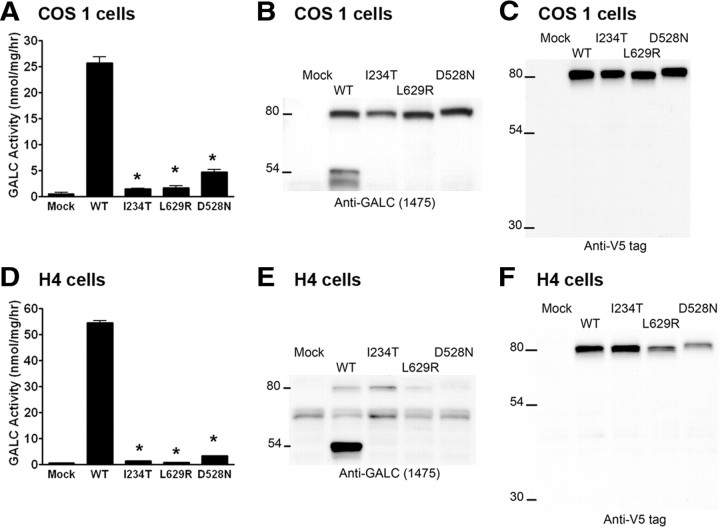
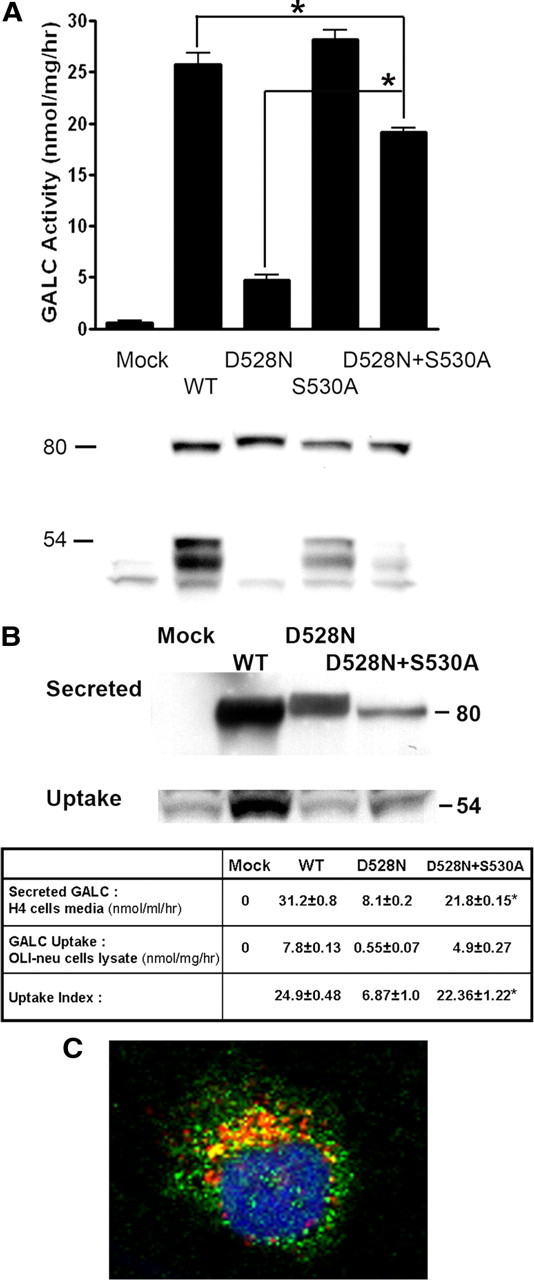
Globoid cell leukodystrophy (GLD) (Krabbe disease) is an autosomal recessive, degenerative, lysosomal storage disease caused by a severe loss of galactocerebrosidase (GALC) enzymatic activity. Of the >70 disease-causing mutations in the GALC gene, most are located outside of the catalytic domain of the enzyme. To determine how GALC mutations impair enzymatic activity, we investigated the impact of multiple disease-causing mutations on GALC processing, localization, and enzymatic activity. Studies in mammalian cells revealed dramatic decreases in GALC activity and a lack of appropriate protein processing into an N-terminal GALC fragment for each of the mutants examined. Consistent with this, we observed significantly less GALC localized to the lysosome and impairment in either the secretion or reuptake of mutant GALC. Notably, the D528N mutation was found to induce hyperglycosylation and protein misfolding. Reversal of these conditions resulted in an increase in proper processing and GALC activity, suggesting that glycosylation may play a critical role in the disease process in patients with this mutation. Recent studies have shown that enzyme inhibitors can sometimes "chaperone" misfolded polypeptides to their appropriate target organelle, bypassing the normal cellular quality control machinery and resulting in enhanced activity. To determine whether this may also work for GLD, we examined the effect of alpha-lobeline, an inhibitor of GALC, on D528N mutant cells. After treatment, GALC activity was significantly increased. This study suggests that mutations in GALC can cause GLD by impairing protein processing and/or folding and that pharmacological chaperones may be potential therapeutic agents for patients carrying certain mutations.
Figures






References
-
- Berson JF, Frank DW, Calvo PA, Bieler BM, Marks MS. A common temperature-sensitive allelic form of human tyrosinase is retained in the endoplasmic reticulum at the nonpermissive temperature. J Biol Chem. 2000;275:12281–12289. - PubMed
-
- Butters TD. Gaucher disease. Curr Opin Chem Biol. 2007;11:412–418. - PubMed
-
- Chen YQ, Wenger DA. Galactocerebrosidase from human urine: purification and partial characterization. Biochim Biophys Acta. 1993;1170:53–61. - PubMed
-
- Chen YQ, Rafi MA, de Gala G, Wenger DA. Cloning and expression of cDNA encoding human galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum Mol Genet. 1993;2:1841–1845. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical