

XPA impacts formation but not proteasome-sensitive repair of DNA-protein cross-links induced by chromate
- PMID: 20410141
- PMCID: PMC2893307
- DOI: 10.1093/mutage/geq017
XPA impacts formation but not proteasome-sensitive repair of DNA-protein cross-links induced by chromate
Abstract
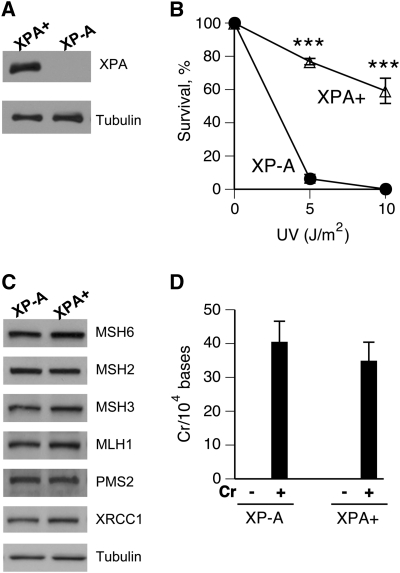
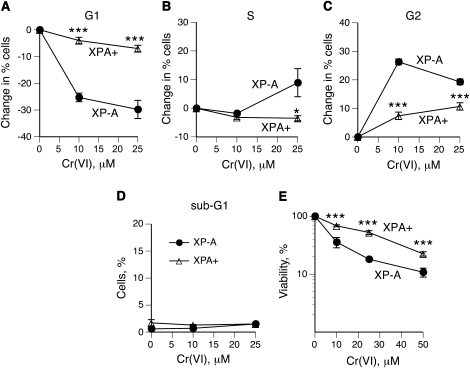
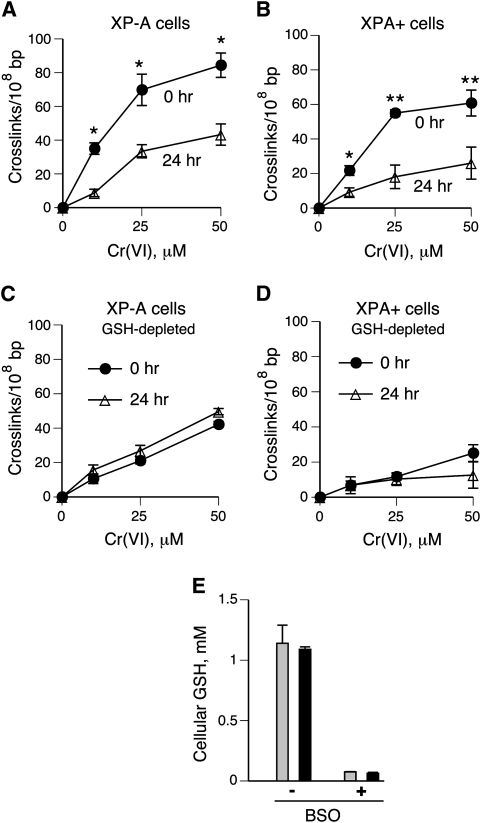
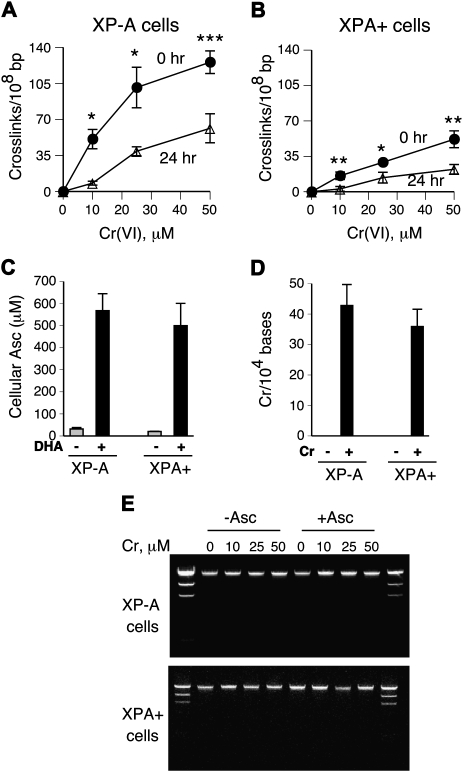
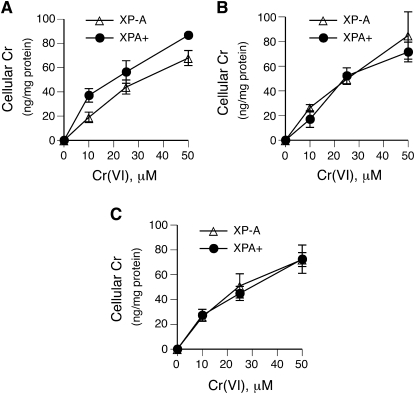
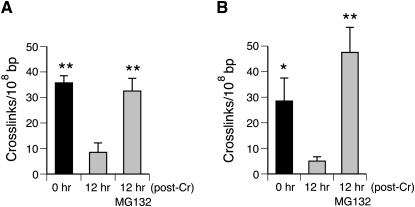
DNA-protein cross-links (DPCs) are caused by a large number of human carcinogens and anti-cancer drugs. However, cellular processes involved in decreasing a burden of these genotoxic lesions remain poorly understood. Here, we examined the impact of nucleotide excision repair (NER), which is a principal repair pathway for bulky DNA adducts, and the main cellular reducers on removal of chromium(VI)-induced DPC. We found that standard and ascorbate-restored cultures of isogenic XPA-null (NER deficient) and XPA-complemented human fibroblasts had very similar repair of Cr-DPC (60-65% average DPC removal after 24 h). However, XPA absence caused depletion of G1 and accumulation of G2 cells at low Cr(VI) doses, suggesting that Cr-DPC were not a significant cause of cell cycle perturbations. Interestingly, although pro-oxidant metabolism of Cr(VI) in glutathione-depleted cells generated significantly fewer DPC, they were repair resistant irrespective of the NER status of cells. Inhibition of proteasome activity by MG132 abolished DPC repair in both XPA-null and XPA-complemented cells. XPA loss caused two to three times higher initial DPC formation, demonstrating the importance of NER in removal of the precursor lesions. Our results indicate that human NER is not involved in removal of Cr-DPC containing non-histone proteins but it acts as a defence mechanism against these large lesions by preventing their formation. Therefore, individual differences in NER activity are expected to alter sensitivity but not persistence of DPC as a biomarker of hexavalent Cr.
Figures
References
-
- Costa M, Zhitkovich A, Harris M, Paustenbauch D, Gargas M. DNA-protein crosslinks produced by various chemicals in cultured human lymphoma cells. J. Toxicol. Environ. Health. 1997;50:433–449. - PubMed
-
- Taioli E, Zhitkovich A, Toniolo P, Bernstein J, Blum R, Costa M. DNA-protein crosslinks as a biomarker of cis-platinum activity in cancer patients. Oncol. Rep. 1996;3:439–441. - PubMed
-
- Barker S, Weinfeld M, Murray D. DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat. Res. 2005;589:111–135. - PubMed
