

Virus inhibition of RIP3-dependent necrosis
- PMID: 20413098
- PMCID: PMC4279434
- DOI: 10.1016/j.chom.2010.03.006
Virus inhibition of RIP3-dependent necrosis
Abstract
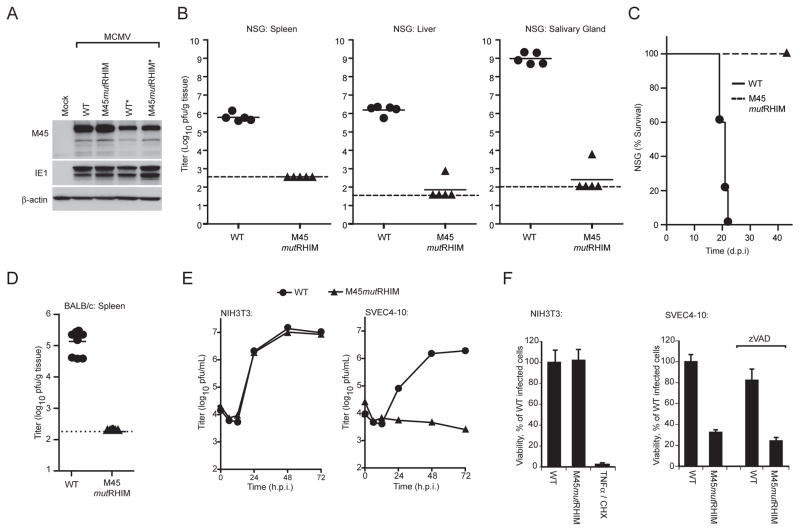
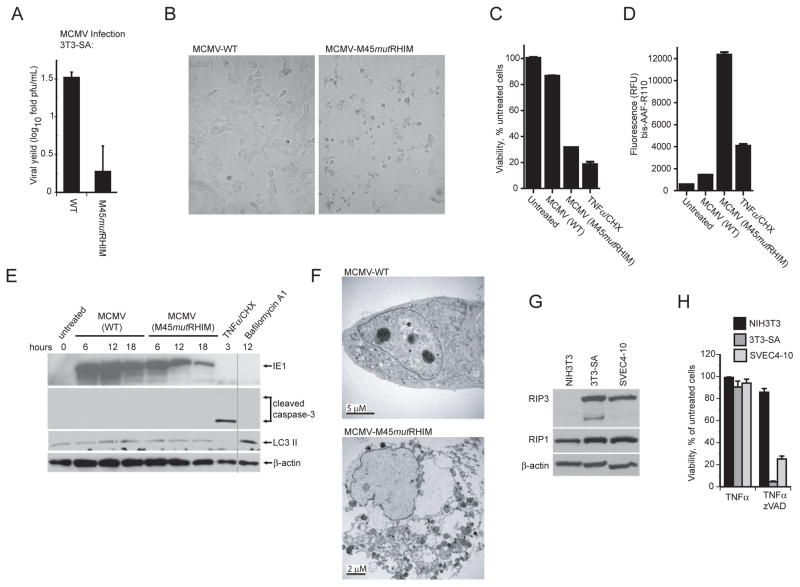
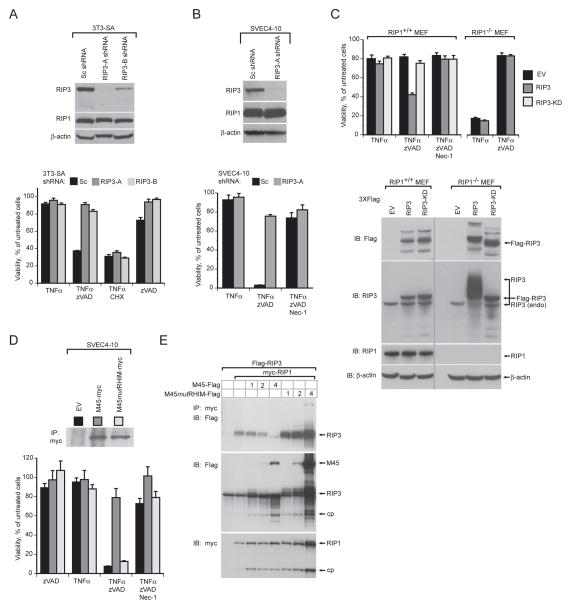
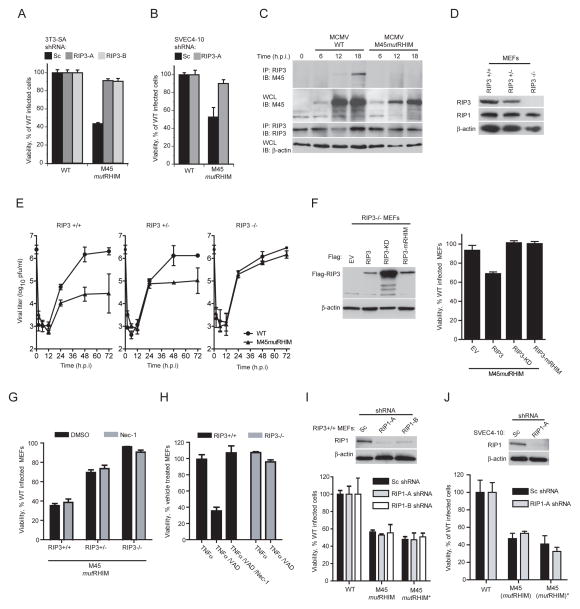
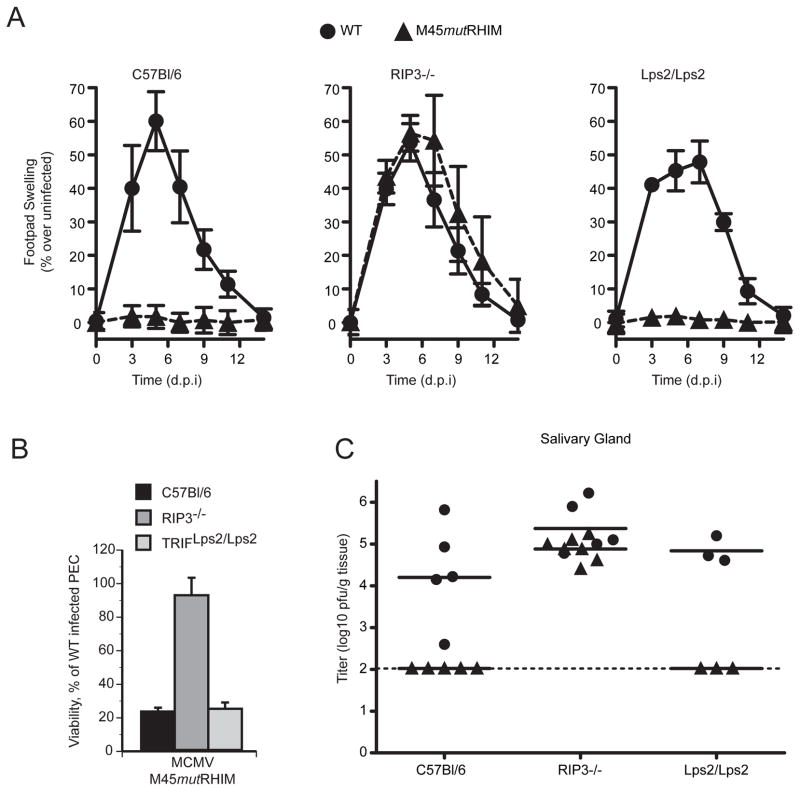
Viral infection activates cytokine expression and triggers cell death, the modulation of which is important for successful pathogenesis. Necroptosis is a form of programmed necrosis dependent on two related RIP homotypic interaction motif (RHIM)-containing signaling adaptors, receptor-interacting protein kinases (RIP) 1 and 3. We find that murine cytomegalovirus infection induces RIP3-dependent necrosis. Whereas RIP3 kinase activity and RHIM-dependent interactions control virus-associated necrosis, virus-induced death proceeds independently of RIP1 and is therefore distinct from TNFalpha-dependent necroptosis. Viral M45-encoded inhibitor of RIP activation (vIRA) targets RIP3 during infection and disrupts RIP3-RIP1 interactions characteristic of TNFalpha-induced necroptosis, thereby suppressing both death pathways. Importantly, attenuation of vIRA mutant virus in wild-type mice is normalized in RIP3-deficient mice. Thus, vIRA function validates necrosis as central to host defense against viral infections and highlights the benefit of multiple virus-encoded cell-death suppressors that inhibit not only apoptotic, but also necrotic mechanisms of virus clearance.
Copyright 2010 Elsevier Inc. All rights reserved.
Figures
References
-
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. - PubMed
-
- Benedict CA. Viruses and the TNF-related cytokines, an evolving battle. Cytokine Growth Factor Rev. 2003;14:349–357. - PubMed
-
- Benedict CA, Banks TA, Ware CF. Death and survival: viral regulation of TNF signaling pathways. Curr Opin Immunol. 2003;15:59–65. - PubMed
-
- Brune W, Menard C, Heesemann J, Koszinowski UH. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science. 2001;291:303–305. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
