

Role of ATM and the damage response mediator proteins 53BP1 and MDC1 in the maintenance of G(2)/M checkpoint arrest
- PMID: 20421415
- PMCID: PMC2897583
- DOI: 10.1128/MCB.01644-09
Role of ATM and the damage response mediator proteins 53BP1 and MDC1 in the maintenance of G(2)/M checkpoint arrest
Abstract
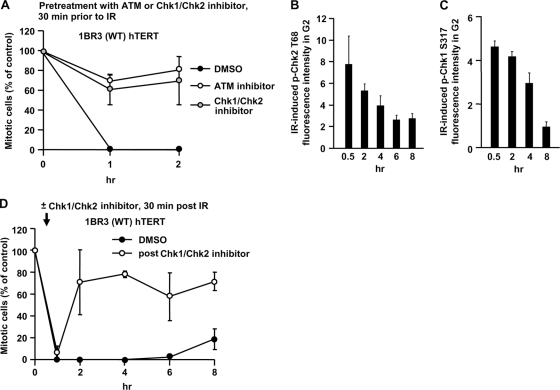
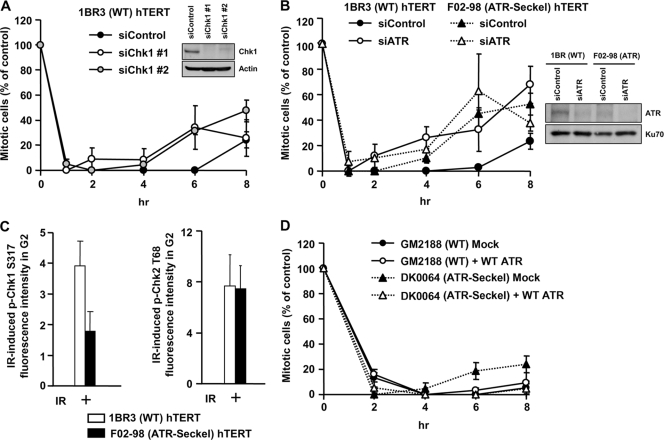
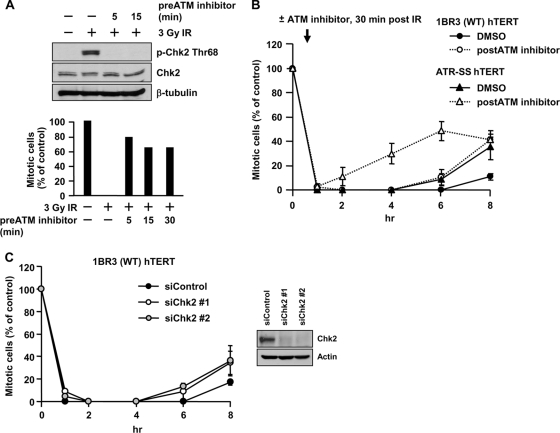
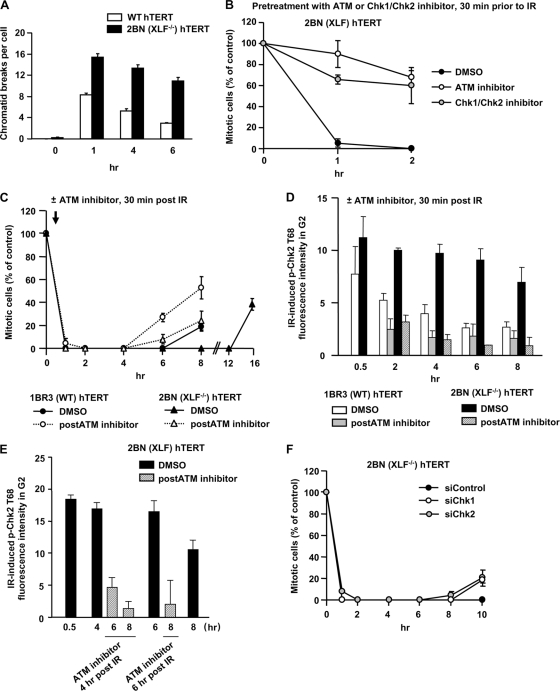
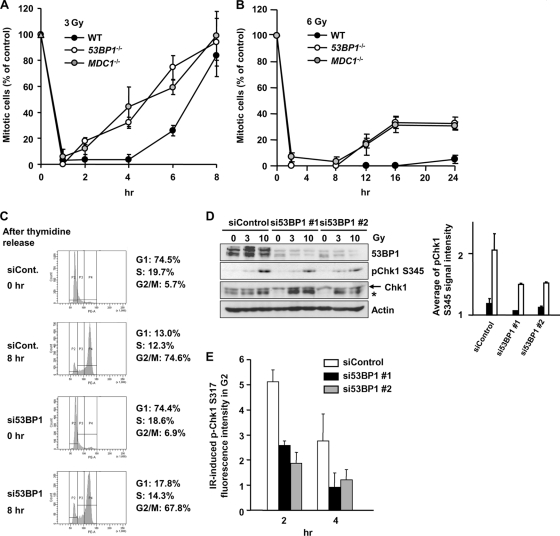
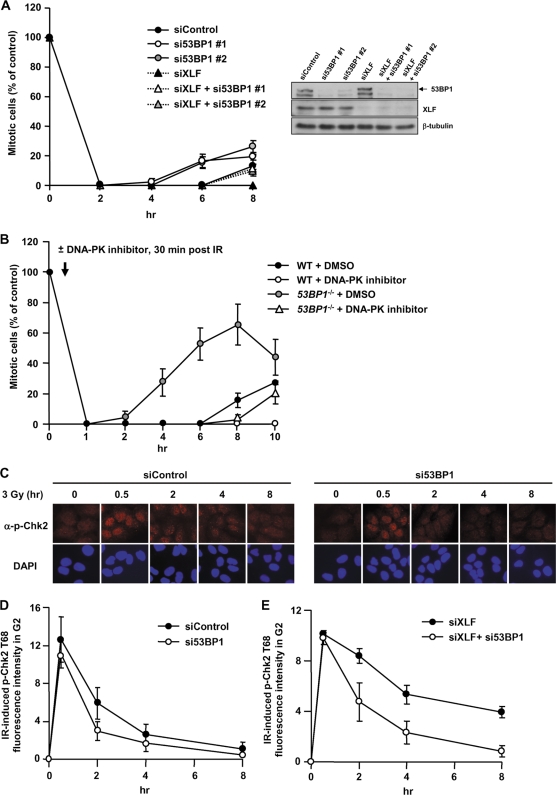
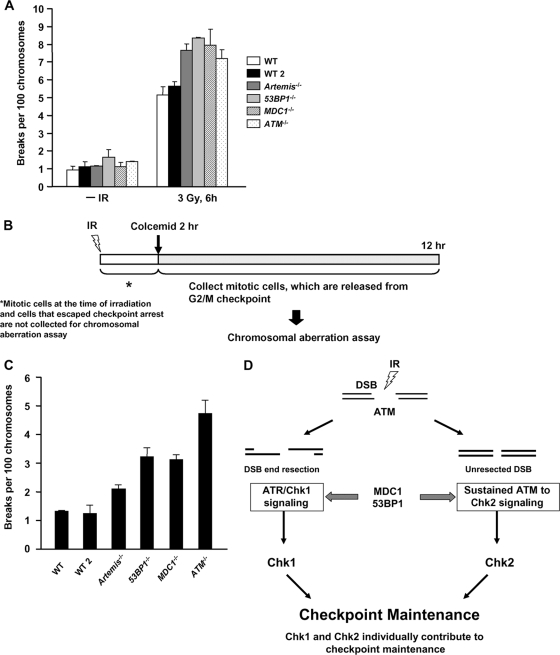
ATM-dependent initiation of the radiation-induced G(2)/M checkpoint arrest is well established. Recent results have shown that the majority of DNA double-strand breaks (DSBs) in G(2) phase are repaired by DNA nonhomologous end joining (NHEJ), while approximately 15% of DSBs are slowly repaired by homologous recombination. Here, we evaluate how the G(2)/M checkpoint is maintained in irradiated G(2) cells, in light of our current understanding of G(2) phase DSB repair. We show that ATM-dependent resection at a subset of DSBs leads to ATR-dependent Chk1 activation. ATR-Seckel syndrome cells, which fail to efficiently activate Chk1, and small interfering RNA (siRNA) Chk1-treated cells show premature mitotic entry. Thus, Chk1 significantly contributes to maintaining checkpoint arrest. Second, sustained ATM signaling to Chk2 contributes, particularly when NHEJ is impaired by XLF deficiency. We also show that cells lacking the mediator proteins 53BP1 and MDC1 initially arrest following radiation doses greater than 3 Gy but are subsequently released prematurely. Thus, 53BP1(-/-) and MDC1(-/-) cells manifest a checkpoint defect at high doses. This failure to maintain arrest is due to diminished Chk1 activation and a decreased ability to sustain ATM-Chk2 signaling. The combined repair and checkpoint defects conferred by 53BP1 and MDC1 deficiency act synergistically to enhance chromosome breakage.
Figures
Similar articles
-
Artemis is a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damage checkpoint response.Mol Cell Biol. 2004 Oct;24(20):9207-20. doi: 10.1128/MCB.24.20.9207-9220.2004. Mol Cell Biol. 2004. PMID: 15456891 Free PMC article.
-
STAT-1 facilitates the ATM activated checkpoint pathway following DNA damage.J Cell Sci. 2005 Apr 15;118(Pt 8):1629-39. doi: 10.1242/jcs.01728. Epub 2005 Mar 22. J Cell Sci. 2005. Retraction in: J Cell Sci. 2015 Mar 1;128(5):1064. doi: 10.1242/jcs.169243. PMID: 15784679 Retracted.
-
The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax.DNA Repair (Amst). 2010 Dec 10;9(12):1273-82. doi: 10.1016/j.dnarep.2010.09.013. Epub 2010 Oct 30. DNA Repair (Amst). 2010. PMID: 21036673 Review.
-
53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage.Cancer Res. 2003 Dec 15;63(24):8586-91. Cancer Res. 2003. PMID: 14695167
-
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer.Adv Cancer Res. 2010;108:73-112. doi: 10.1016/B978-0-12-380888-2.00003-0. Adv Cancer Res. 2010. PMID: 21034966 Review.
Cited by
-
A role for homologous recombination proteins in cell cycle regulation.Cell Cycle. 2015;14(17):2853-61. doi: 10.1080/15384101.2015.1049784. Epub 2015 Jun 30. Cell Cycle. 2015. PMID: 26125600 Free PMC article.
-
CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1.Nucleic Acids Res. 2011 Mar;39(6):2144-52. doi: 10.1093/nar/gkq1175. Epub 2010 Nov 17. Nucleic Acids Res. 2011. PMID: 21087997 Free PMC article.
-
The Role of Nitric Oxide in Cancer: Master Regulator or NOt?Int J Mol Sci. 2020 Dec 10;21(24):9393. doi: 10.3390/ijms21249393. Int J Mol Sci. 2020. PMID: 33321789 Free PMC article. Review.
-
Monitoring the Activation of the DNA Damage Response Pathway in a 3D Spheroid Model.PLoS One. 2015 Jul 30;10(7):e0134411. doi: 10.1371/journal.pone.0134411. eCollection 2015. PLoS One. 2015. PMID: 26225756 Free PMC article.
-
The 53BP1 homolog in C. elegans influences DNA repair and promotes apoptosis in response to ionizing radiation.PLoS One. 2013 May 8;8(5):e64028. doi: 10.1371/journal.pone.0064028. Print 2013. PLoS One. 2013. PMID: 23667696 Free PMC article.
References
-
- Ahnesorg, P., P. Smith, and S. P. Jackson. 2006. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124:301-313. - PubMed
-
- Beamish, H., and M. F. Lavin. 1994. Radiosensitivity in ataxia-telangiectasia: anomalies in radiation-induced cell cycle delay. Int. J. Radiat. Biol. 65:175-184. - PubMed
-
- Buck, D., L. Malivert, R. de Chasseval, A. Barraud, M. C. Fondaneche, O. Sanal, A. Plebani, J. L. Stephan, M. Hufnagel, F. le Deist, A. Fischer, A. Durandy, J. P. de Villartay, and P. Revy. 2006. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124:287-299. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous