

Methylenedioxymethamphetamine inhibits mitochondrial complex I activity in mice: a possible mechanism underlying neurotoxicity
- PMID: 20423338
- PMCID: PMC2874846
- DOI: 10.1111/j.1476-5381.2010.00663.x
Methylenedioxymethamphetamine inhibits mitochondrial complex I activity in mice: a possible mechanism underlying neurotoxicity
Abstract
Background and purpose: 3,4-methylenedioxymethamphetamine (MDMA) causes a persistent loss of dopaminergic cell bodies in the substantia nigra of mice. Current evidence indicates that such neurotoxicity is due to oxidative stress but the source of free radicals remains unknown. Inhibition of mitochondrial electron transport chain complexes by MDMA was assessed as a possible source.
Experimental approach: Activities of mitochondrial complexes after MDMA were evaluated spectrophotometrically. In situ visualization of superoxide production in the striatum was assessed by ethidium fluorescence and striatal dopamine levels were determined by HPLC as an index of dopaminergic toxicity.
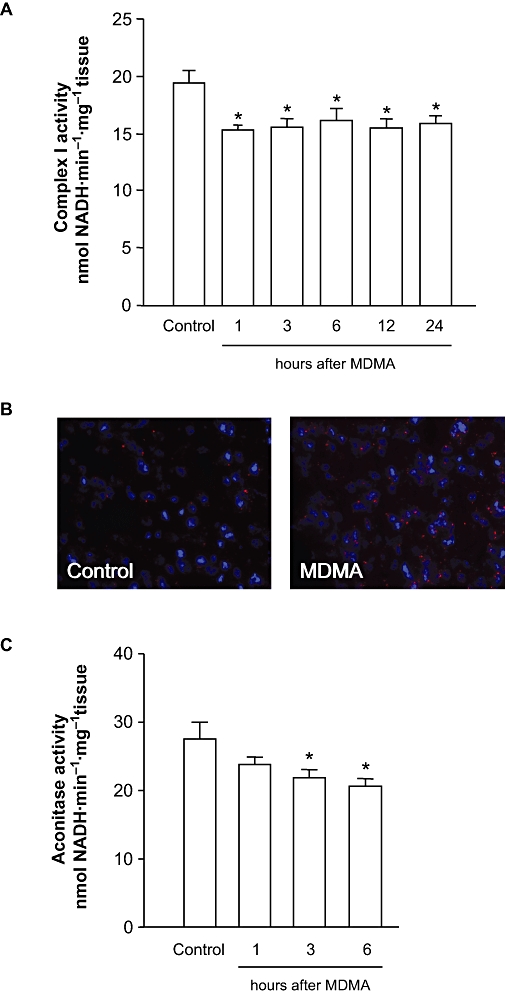
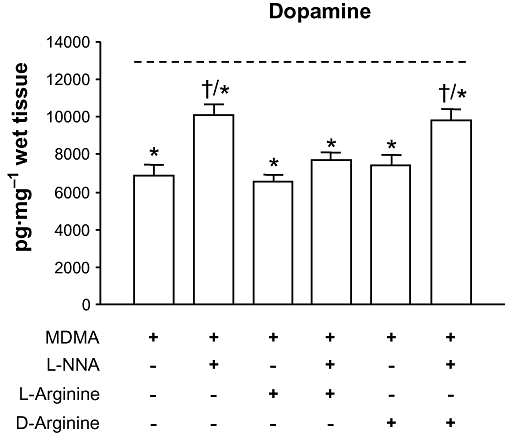
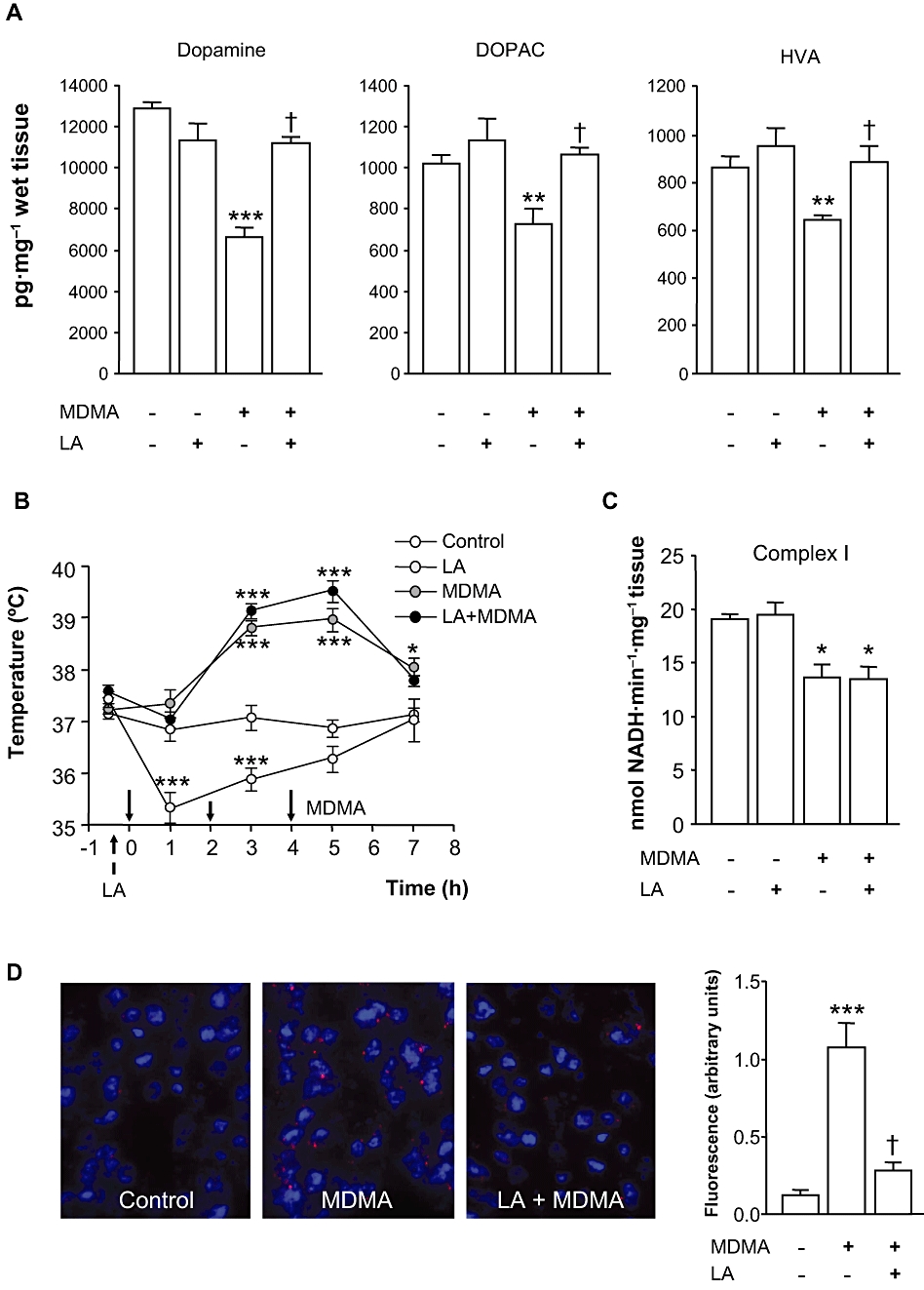
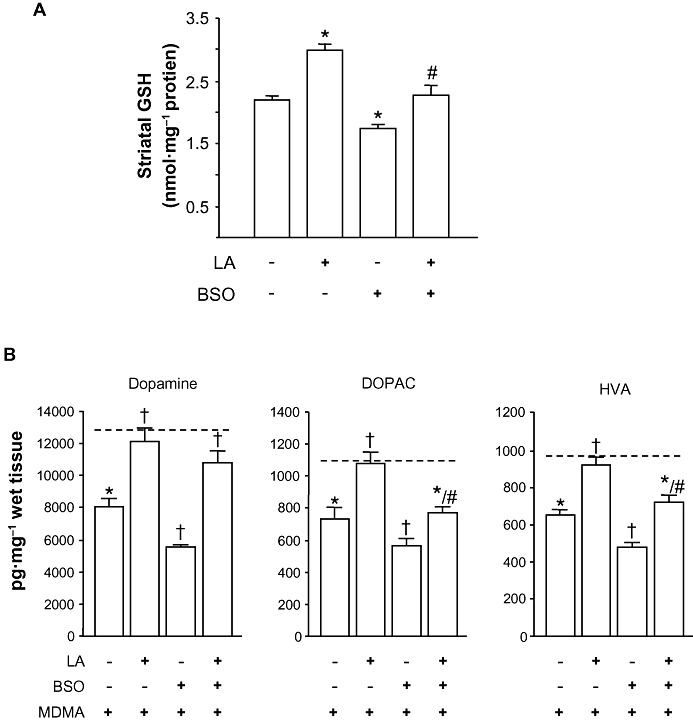
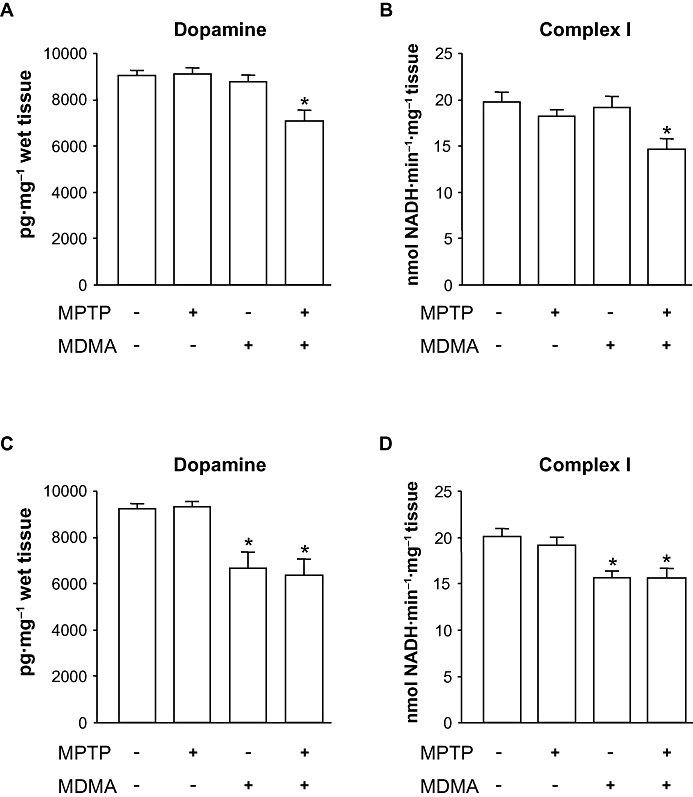
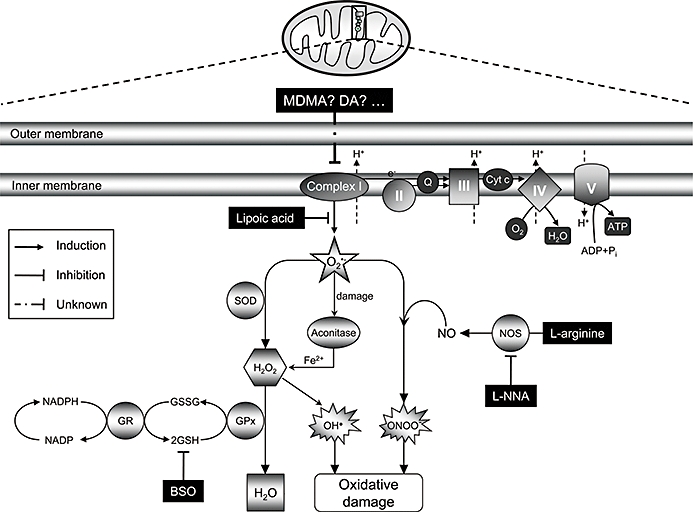
Key results: 3,4-methylenedioxymethamphetamine decreased mitochondrial complex I activity in the striatum of mice, an effect accompanied by an increased production of superoxide radicals and the inhibition of endogenous aconitase. alpha-Lipoic acid prevented superoxide generation and long-term toxicity independent of any effect on complex I inhibition. These effects of alpha-lipoic acid were also associated with a significant increase of striatal glutathione levels. The relevance of glutathione was supported by reducing striatal glutathione content with L-buthionine-(S,R)-sulfoximine, which exacerbated MDMA-induced dopamine deficits, effects suppressed by alpha-lipoic acid. The nitric oxide synthase inhibitor, N(G)-nitro-L-arginine, partially prevented MDMA-induced dopamine depletions, an effect reversed by L-arginine but not D-arginine. Finally, a direct relationship between mitochondrial complex I inhibition and long-term dopamine depletions was found in animals treated with MDMA in combination with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
Conclusions and implications: Inhibition of mitochondrial complex I following MDMA could be the source of free radicals responsible for oxidative stress and the consequent neurotoxicity of this drug in mice.
Figures
Comment in
-
Mitochondria as pharmacological targets.Br J Pharmacol. 2010 May;160(2):217-9. doi: 10.1111/j.1476-5381.2010.00706.x. Br J Pharmacol. 2010. PMID: 20423336 Free PMC article.
References
-
- Aguirre N, Barrionuevo M, Ramírez MJ, Del Río J, Lasheras B. Alpha-lipoic acid prevents 3,4-methylenedioxy-methamphetamine (MDMA)-induced neurotoxicity. Neuroreport. 1999;10:3675–3680. - PubMed
-
- Albers DS, Zeevalk GD, Sonsalla PK. Damage to dopaminergic nerve terminals in mice by combined treatment of intrastriatal malonate with systemic methamphetamine or MPTP. Brain Res. 1996;718:217–220. - PubMed
-
- Aleardi AM, Benard G, Augereau O, Malgat M, Talbot JC, Mazat JP, et al. Gradual alteration of mitochondrial structure and function by beta-amyloids: importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J Bioenerg Biomembr. 2005;37:207–225. - PubMed
-
- Alves E, Summavielle T, Alves CJ, Custódio JB, Fernandes E, de Lourdes Bastos M, et al. Ecstasy-induced oxidative stress to adolescent rat brain mitochondria in vivo: influence of monoamine oxidase type A. Addict Biol. 2009;14:185–193. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
