

Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice
- PMID: 20424327
- PMCID: PMC2860913
- DOI: 10.1172/JCI40266
Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice
Abstract
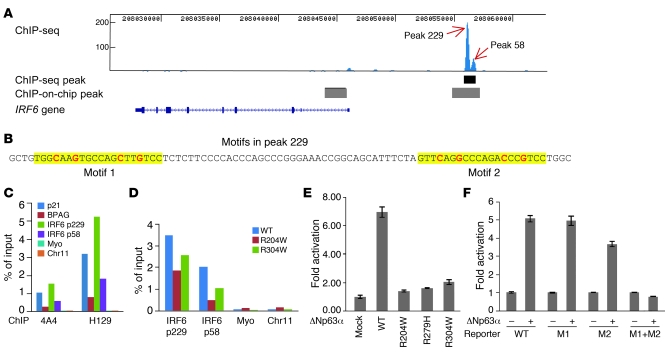
Cleft palate is a common congenital disorder that affects up to 1 in 2,500 live human births and results in considerable morbidity to affected individuals and their families. The etiology of cleft palate is complex, with both genetic and environmental factors implicated. Mutations in the transcription factor-encoding genes p63 and interferon regulatory factor 6 (IRF6) have individually been identified as causes of cleft palate; however, a relationship between the key transcription factors p63 and IRF6 has not been determined. Here, we used both mouse models and human primary keratinocytes from patients with cleft palate to demonstrate that IRF6 and p63 interact epistatically during development of the secondary palate. Mice simultaneously carrying a heterozygous deletion of p63 and the Irf6 knockin mutation R84C, which causes cleft palate in humans, displayed ectodermal abnormalities that led to cleft palate. Furthermore, we showed that p63 transactivated IRF6 by binding to an upstream enhancer element; genetic variation within this enhancer element is associated with increased susceptibility to cleft lip. Our findings therefore identify p63 as a key regulatory molecule during palate development and provide a mechanism for the cooperative role of p63 and IRF6 in orofacial development in mice and humans.
Figures
Comment in
-
p63 and IRF6: brothers in arms against cleft palate.J Clin Invest. 2010 May;120(5):1386-9. doi: 10.1172/JCI42821. Epub 2010 Apr 26. J Clin Invest. 2010. PMID: 20424318 Free PMC article.
References
-
- Vanderas AP. Incidence of cleft lip, cleft palate, and cleft lip and palate among races: a review. Cleft Palate J. 1987;24(3):216–225. - PubMed
-
- Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. 2009;374(9703):1773–1785. - PubMed
-
- Jones MC. Etiology of facial clefts: prospective evaluation of 428 patients. Cleft Palate J. 1988;25(1):16–20. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
