

The role of MeCP2 in brain development and neurodevelopmental disorders
- PMID: 20425298
- PMCID: PMC2847695
- DOI: 10.1007/s11920-010-0097-7
The role of MeCP2 in brain development and neurodevelopmental disorders
Abstract
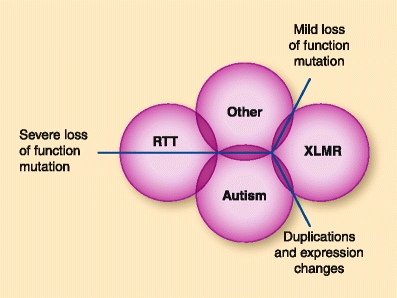
Methyl CpG binding protein-2 (MeCP2) is an essential epigenetic regulator in human brain development. Rett syndrome, the primary disorder caused by mutations in the X-linked MECP2 gene, is characterized by a period of cognitive decline and development of hand stereotypies and seizures following an apparently normal early infancy. In addition, MECP2 mutations and duplications are observed in a spectrum of neurodevelopmental disorders, including severe neonatal encephalopathy, X-linked mental retardation, and autism, implicating MeCP2 as an essential regulator of postnatal brain development. In this review, we compare the mutation types and inheritance patterns of the human disorders associated with MECP2. In addition, we summarize the current understanding of MeCP2 as a central epigenetic regulator of activity-dependent synaptic maturation. As MeCP2 occupies a central role in the pathogenesis of multiple neurodevelopmental disorders, continued investigation into MeCP2 function and regulatory pathways may show promise for developing broad-spectrum therapies.
Figures
References
-
- Zoghbi HY. MeCP2 dysfunction in humans and mice. J Child Neurol. 2005;20:736–740. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
