

Signals from dying hepatocytes trigger growth of liver progenitors
- PMID: 20427400
- PMCID: PMC3632642
- DOI: 10.1136/gut.2009.204354
Signals from dying hepatocytes trigger growth of liver progenitors
Abstract
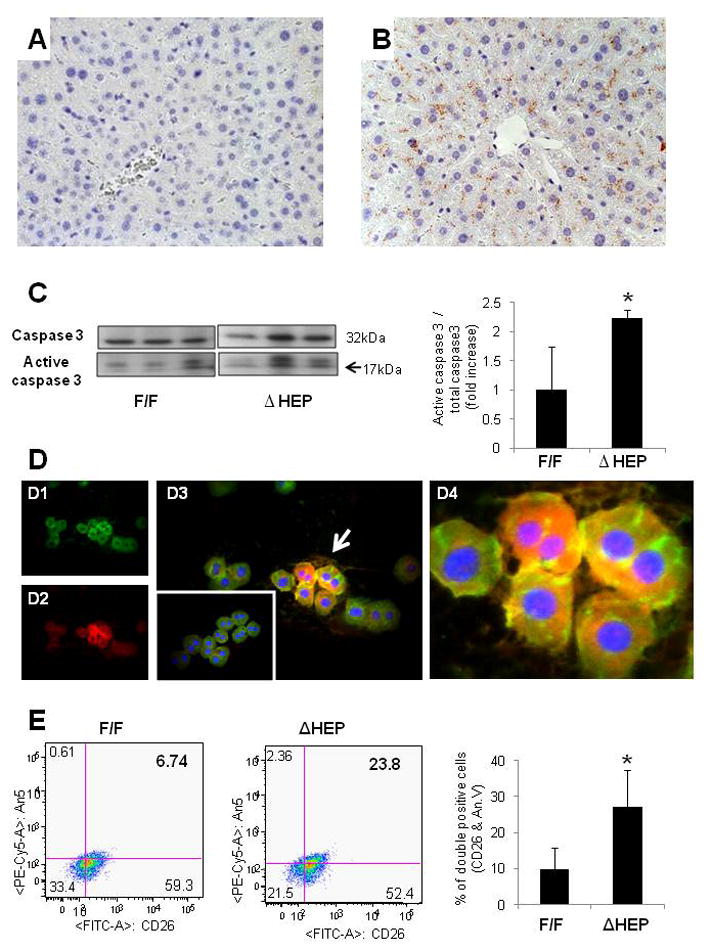
Objective: The death rate of mature hepatocytes is chronically increased in various liver diseases, triggering responses that prevent liver atrophy, but often cause fibrosis. Mice with targeted disruption of inhibitor kappa B kinase (Ikk) in hepatocytes (HEP mice) provide a model to investigate this process because inhibiting Ikk-nuclear factor-kappaB (NF-kappaB) signalling in hepatocytes increases their apoptosis.
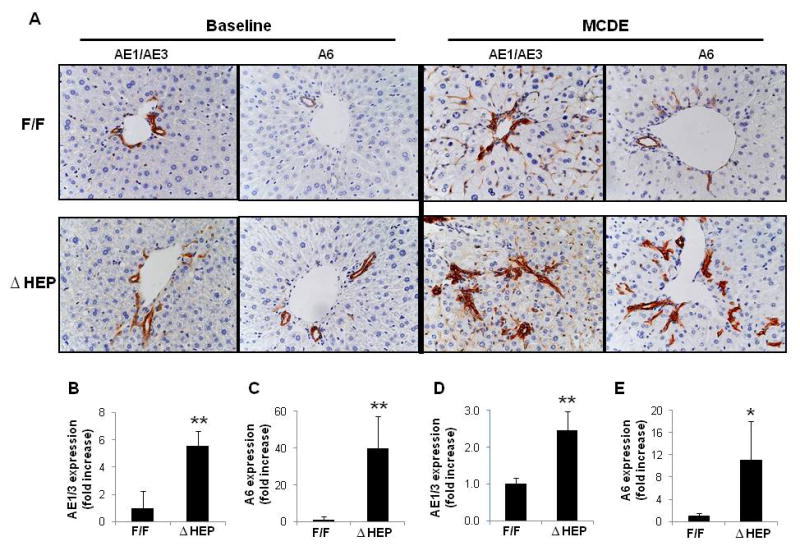
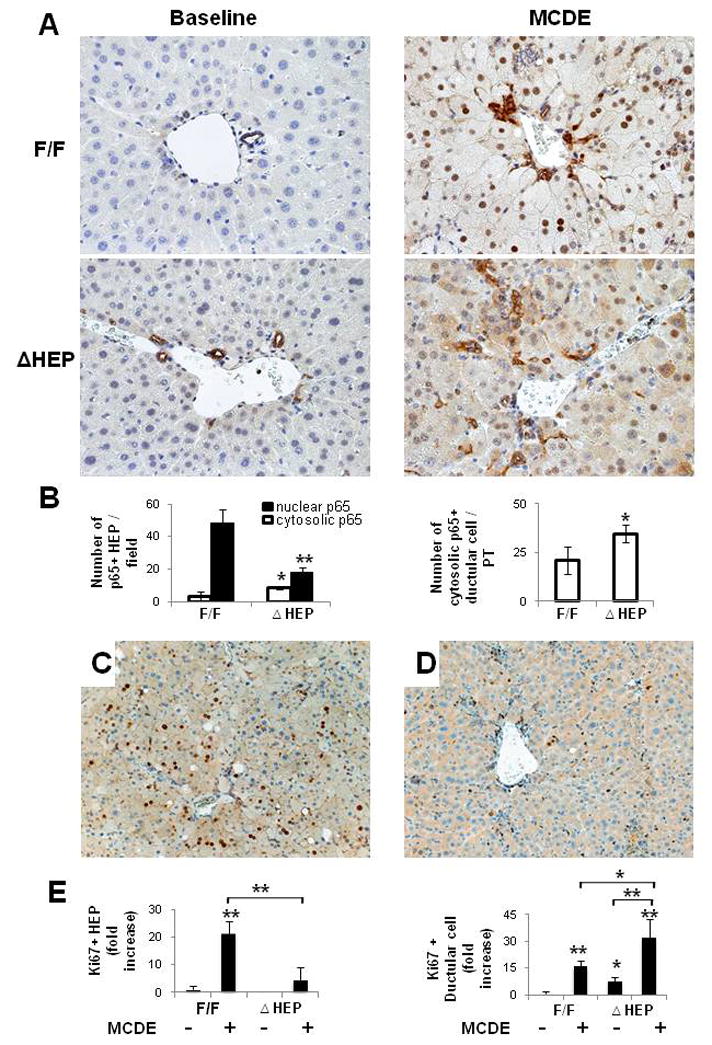
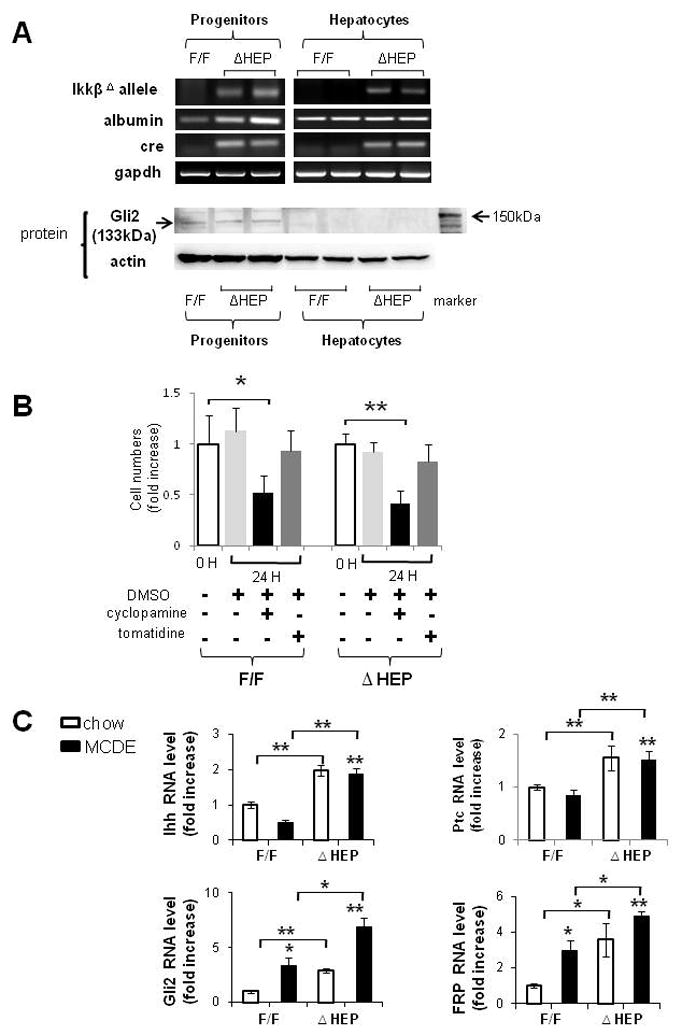
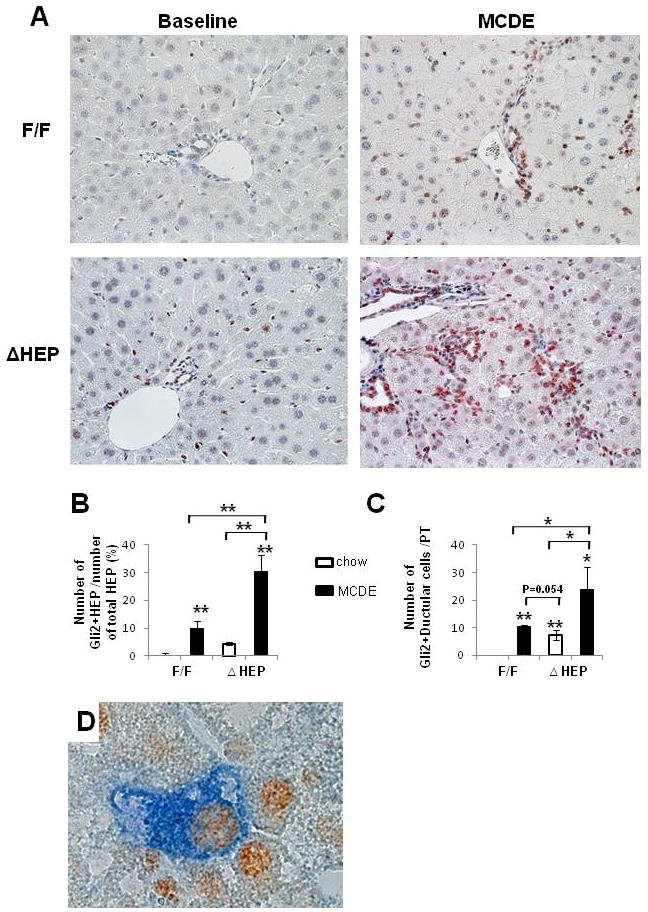
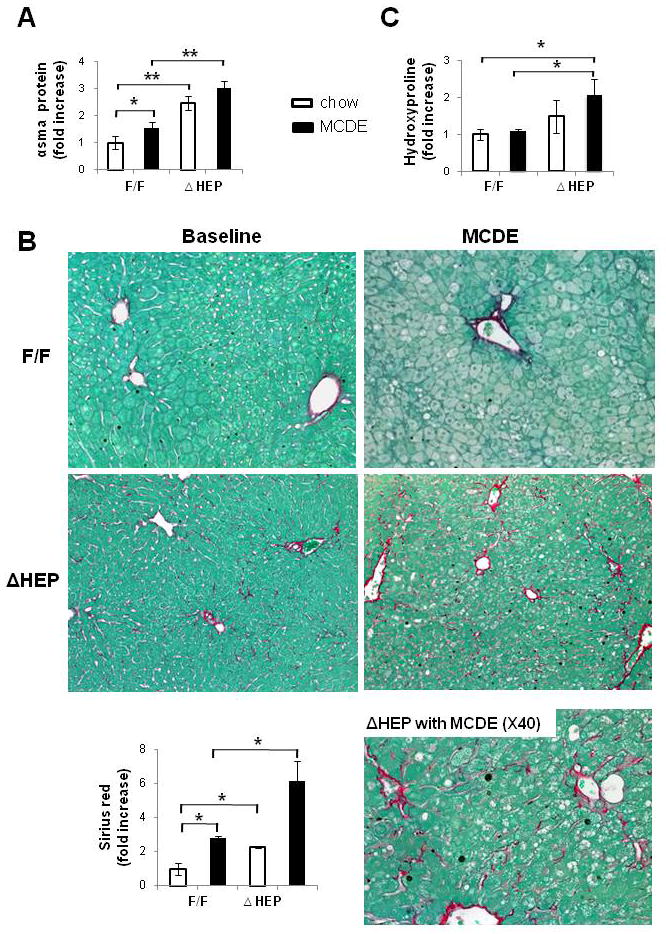
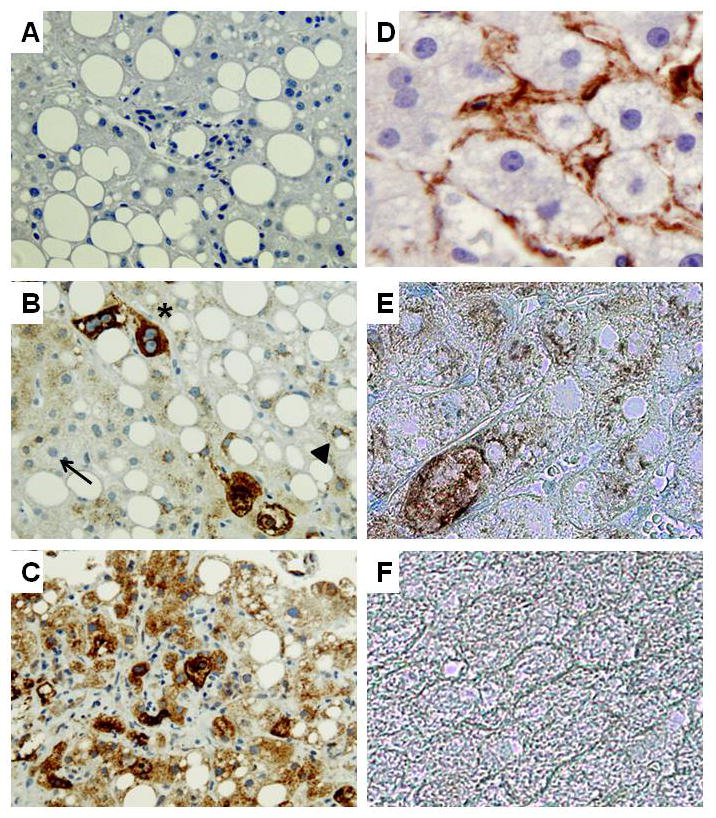
Methods: Cell proliferation, apoptosis, progenitors, fibrosis and production of Hedgehog (Hh) ligands (progenitor and myofibroblast growth factors) were compared in HEP and control mice before and after feeding methionine choline-deficient ethionine-supplemented (MCDE) diets. Ikkbeta was deleted from primary hepatocytes to determine the effects on Hh ligand production; Hh signalling was inhibited directly in progenitors to determine the effects on viability. Liver sections from patients were examined to assess relationships between hepatocyte production of Hh ligands, accumulation of myofibroblastic cells and liver fibrosis.
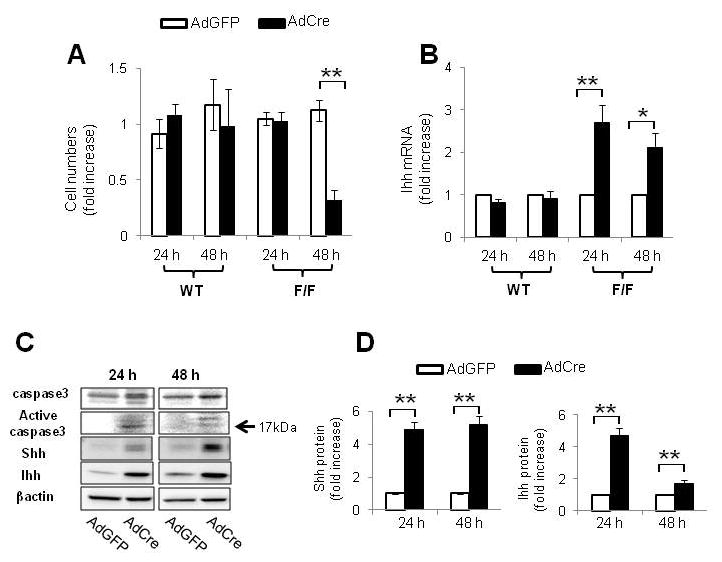
Results: Disrupting the Ikk-NF-kappaB pathway in hepatocytes inhibited their proliferation but induced their production of Hh ligands. The latter provided viability signals for progenitors and myofibroblasts, enhancing accumulation of these cell types and causing fibrogenesis. Findings in the mouse models were recapitulated in diseased human livers.
Conclusion: Dying mature hepatocytes produce Hh ligands which promote the compensatory outgrowth of progenitors and myofibroblasts. These results help to explain why diseases that chronically increase hepatocyte death promote cirrhosis.
Conflict of interest statement
All authors declare that they have no conflict of interests or financial interests
Figures
References
-
- Henderson NC, Forbes SJ. Hepatic fibrogenesis: from within and outwith. Toxicology. 2008;254:130–5. - PubMed
-
- Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–6. - PubMed
-
- Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006:re13. - PubMed
-
- Cosulich SC, James NH, Needham MR, et al. A dominant negative form of IKK2 prevents suppression of apoptosis by the peroxisome proliferator nafenopin. Carcinogenesis. 2000;21:1757–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical