

Parallelization of the MAFFT multiple sequence alignment program
- PMID: 20427515
- PMCID: PMC2905546
- DOI: 10.1093/bioinformatics/btq224
Parallelization of the MAFFT multiple sequence alignment program
Abstract
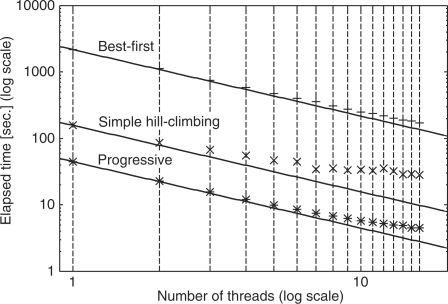
Summary: Multiple sequence alignment (MSA) is an important step in comparative sequence analyses. Parallelization is a key technique for reducing the time required for large-scale sequence analyses. The three calculation stages, all-to-all comparison, progressive alignment and iterative refinement, of the MAFFT MSA program were parallelized using the POSIX Threads library. Two natural parallelization strategies (best-first and simple hill-climbing) were implemented for the iterative refinement stage. Based on comparisons of the objective scores and benchmark scores between the two approaches, we selected a simple hill-climbing approach as the default.
Availability: The parallelized version of MAFFT is available at http://mafft.cbrc.jp/alignment/software/. This version currently supports the Linux operating system only.
Figures
References
-
- Barton GJ, Sternberg MJ. A strategy for the rapid multiple alignment of protein sequences. confidence levels from tertiary structure comparisons. J. Mol. Biol. 1987;198:327–337. - PubMed
-
- Berger MP, Munson PJ. A novel randomized iterative strategy for aligning multiple protein sequences. Comput. Appl. Biosci. 1991;7:479–484. - PubMed
-
- Chaichoompu K, et al. Proceedings 20th IEEE International Parallel & Distributed Processing Symposium. IEEE Computer Society Press; 2006. MT-ClustalW: multithreading multiple sequence alignment; p. 280.
-
- Date S, et al. Multiple alignment of sequences on parallel computers. Comput. Appl. Biosci. 1993;9:397–402. - PubMed
-
- Feng DF, Doolittle RF. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol. 1987;25:351–360. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
