

Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage
- PMID: 20427645
- PMCID: PMC2871011
- DOI: 10.1523/JNEUROSCI.0137-10.2010
Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage
Abstract
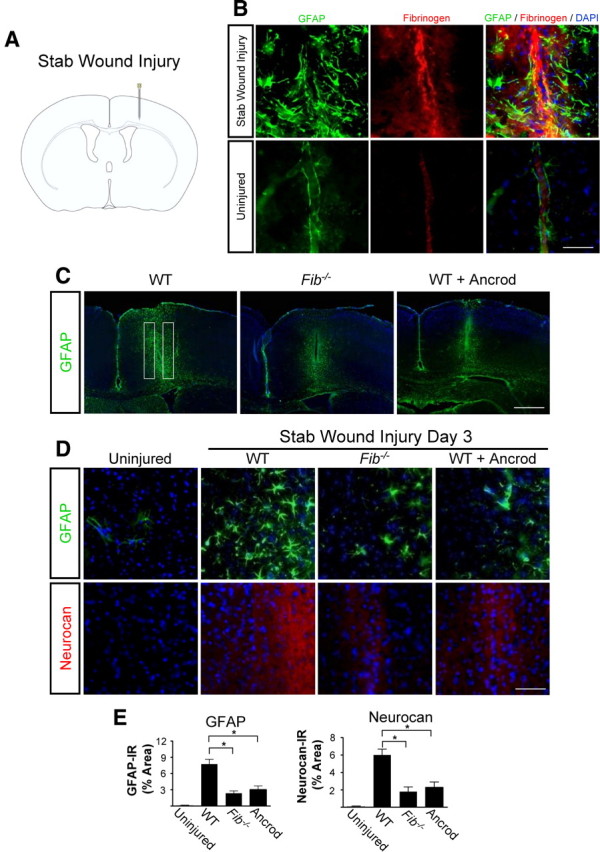
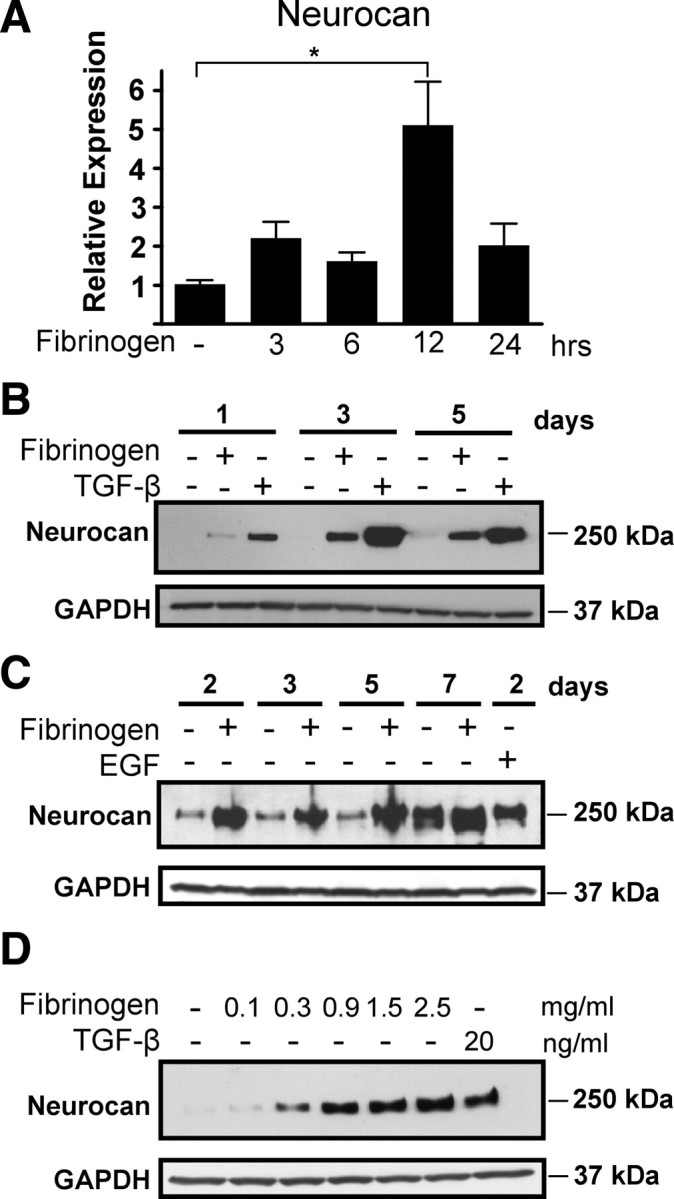
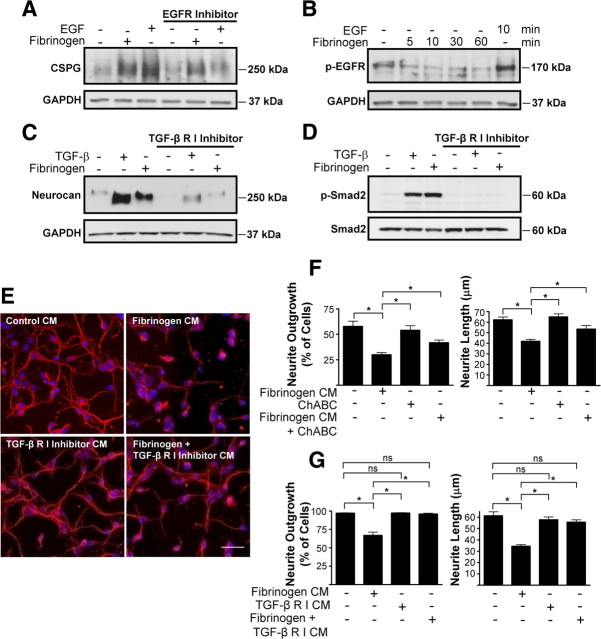
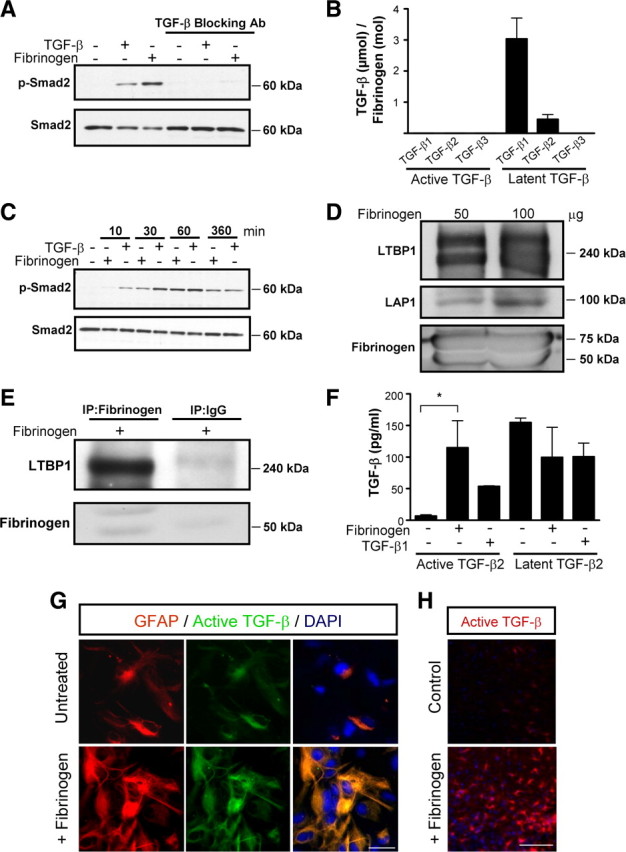
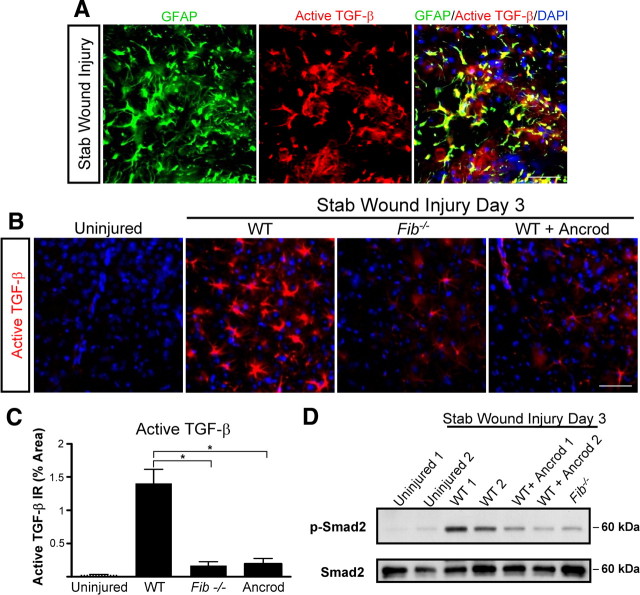
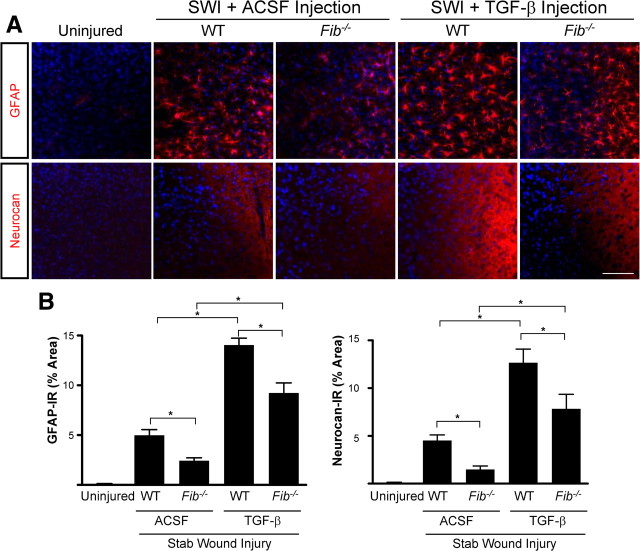
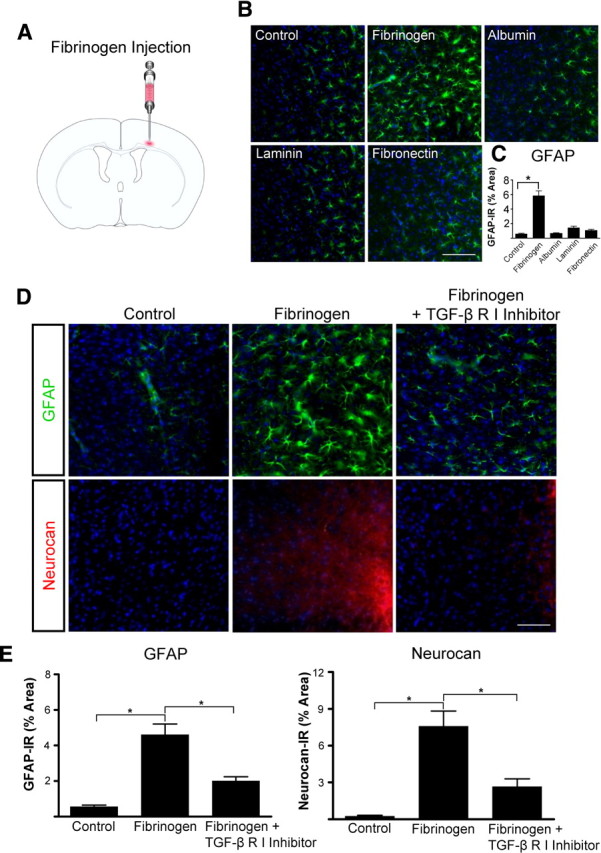
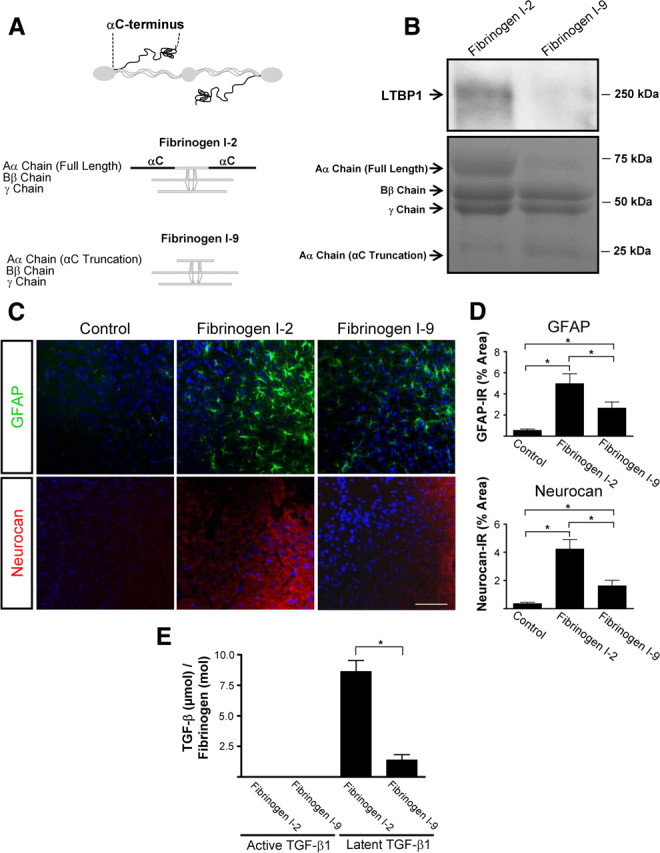
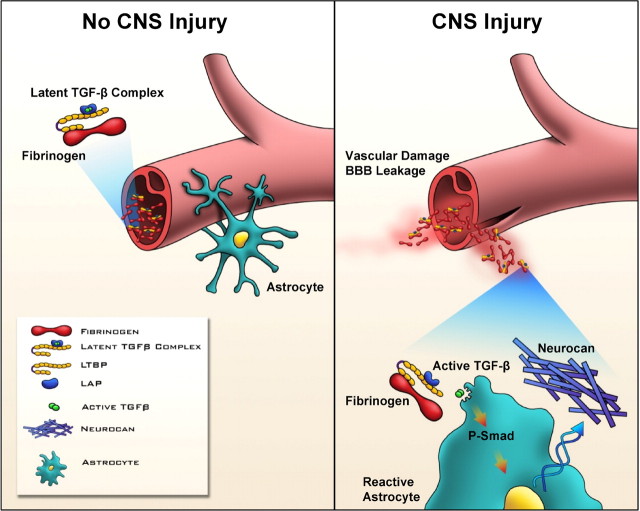
Scar formation in the nervous system begins within hours after traumatic injury and is characterized primarily by reactive astrocytes depositing proteoglycans that inhibit regeneration. A fundamental question in CNS repair has been the identity of the initial molecular mediator that triggers glial scar formation. Here we show that the blood protein fibrinogen, which leaks into the CNS immediately after blood-brain barrier (BBB) disruption or vascular damage, serves as an early signal for the induction of glial scar formation via the TGF-beta/Smad signaling pathway. Our studies revealed that fibrinogen is a carrier of latent TGF-beta and induces phosphorylation of Smad2 in astrocytes that leads to inhibition of neurite outgrowth. Consistent with these findings, genetic or pharmacologic depletion of fibrinogen in mice reduces active TGF-beta, Smad2 phosphorylation, glial cell activation, and neurocan deposition after cortical injury. Furthermore, stereotactic injection of fibrinogen into the mouse cortex is sufficient to induce astrogliosis. Inhibition of the TGF-beta receptor pathway abolishes the fibrinogen-induced effects on glial scar formation in vivo and in vitro. These results identify fibrinogen as a primary astrocyte activation signal, provide evidence that deposition of inhibitory proteoglycans is induced by a blood protein that leaks in the CNS after vasculature rupture, and point to TGF-beta as a molecular link between vascular permeability and scar formation.
Figures
References
-
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. - PubMed
-
- Adams RA, Passino M, Sachs BD, Nuriel T, Akassoglou K. Fibrin mechanisms and functions in nervous system pathology. Mol Interv. 2004;4:163–176. - PubMed
-
- Adams RA, Schachtrup C, Davalos D, Tsigelny I, Akassoglou K. Fibrinogen signal transduction as a mediator and therapeutic target in inflammation: lessons from multiple sclerosis. Curr Med Chem. 2007a;14:2925–2936. - PubMed
-
- Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases