

Potent and use-dependent block of cardiac sodium channels by U-50,488H, a benzeneacetamide kappa opioid receptor agonist
- PMID: 20428265
- PMCID: PMC2859007
Potent and use-dependent block of cardiac sodium channels by U-50,488H, a benzeneacetamide kappa opioid receptor agonist
Abstract
Objectives: To determine whether the kappa opioid receptor agonist U-50,488H, a benzacetamide derivative of the cyclo-hexane-1,2-diamine analgesics, may be a useful molecular probe to define the structural requirements of this class of drugs for cardiac sodium channel blockade.
Animals and methods: The electrophysiological effects of U-50,488H were compared with those of lidocaine, a clinically used class Ib antiarrhythmic agent, in rat heart sodium currents expressed in Xenopus laevis oocytes by using two-electrode voltage clamp.
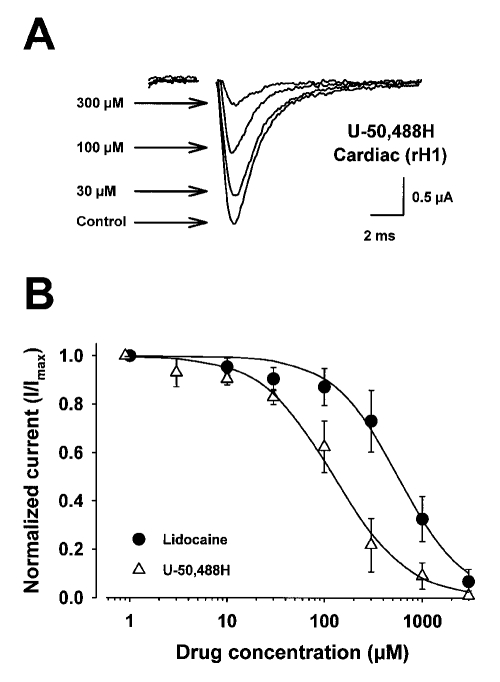
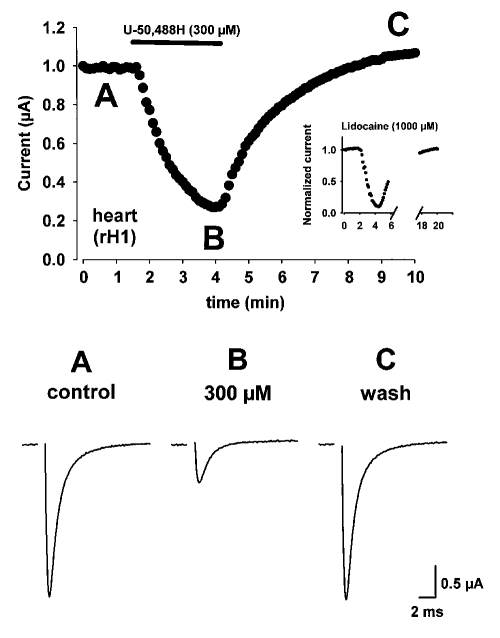
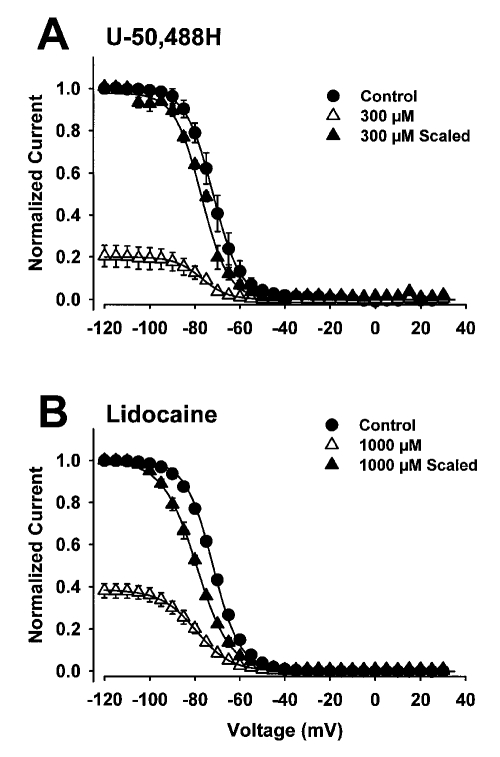
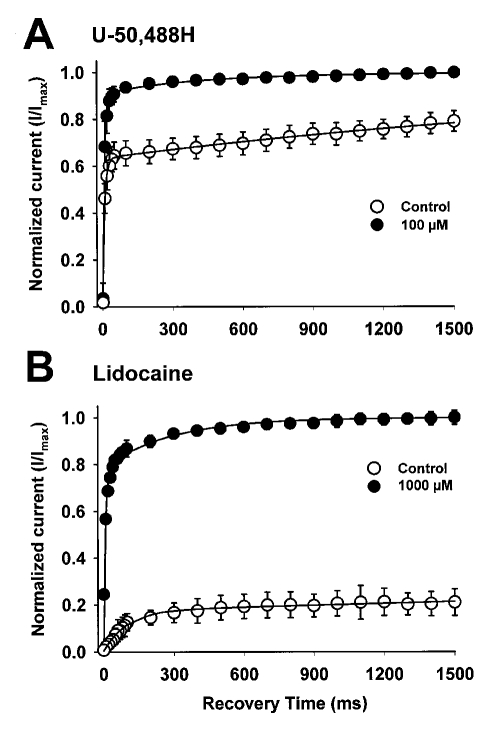
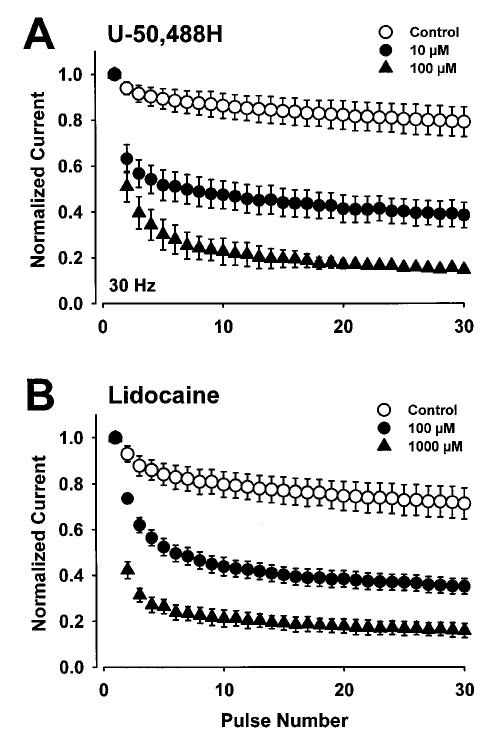
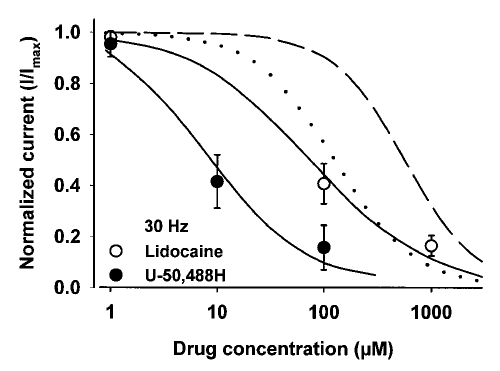
Results: Both U-50,488H and lidocaine produced a concentration-dependent tonic block of sodium current, but U-50,488H was approximately fourfold more potent than lidocaine. Both drugs produced a hyperpolarizing shift in the voltage dependence of sodium channel inactivation and both delayed recovery from inactivation. Both drugs exhibited use-dependent block, but U-50,488H showed a 1.8-fold increase in potency compared with lidocaine at a high frequency of stimulation (30 Hz).
Conclusions: The more potent tonic and use-dependent block of cardiac sodium channels by U-50,488H suggests that structural features of this molecule may provide it with a greater ability to block the channel. An understanding of these structural features may provide information needed in the development of novel arylacetamide-based antiarrhythmic drugs and insight into possible mechanisms describing channel block, resulting in a highly efficacious antiarrhythmic action in the heart.
Keywords: Antiarrhythmic agents; Membrane currents; Sodium channel; U-50,488H.
Figures
Similar articles
-
An electrophysiological basis for the antiarrhythmic actions of the kappa-opioid receptor agonist U-50,488H.Eur J Pharmacol. 1994 Aug 22;261(3):303-9. doi: 10.1016/0014-2999(94)90121-x. Eur J Pharmacol. 1994. PMID: 7813552
-
Effects of bisaramil, a novel class I antiarrhythmic agent, on heart, skeletal muscle and brain Na+ channels.Eur J Pharmacol. 1998 Jan 19;342(1):93-104. doi: 10.1016/s0014-2999(97)01420-9. Eur J Pharmacol. 1998. PMID: 9544797
-
Spiradoline, a kappa opioid receptor agonist, produces tonic- and use-dependent block of sodium channels expressed in Xenopus oocytes.Gen Pharmacol. 2000 Jun;34(6):417-27. doi: 10.1016/s0306-3623(01)00079-9. Gen Pharmacol. 2000. PMID: 11483291
-
U-50,488H, a kappa opioid receptor agonist, is a more potent blocker of cardiac sodium channels than lidocaine.Proc West Pharmacol Soc. 2000;43:47-50. Proc West Pharmacol Soc. 2000. PMID: 11056955 No abstract available.
-
Cardiovascular actions of the kappa-agonist, U-50,488H, in the absence and presence of opioid receptor blockade.Br J Pharmacol. 1992 Mar;105(3):521-6. doi: 10.1111/j.1476-5381.1992.tb09012.x. Br J Pharmacol. 1992. PMID: 1320979 Free PMC article.
Cited by
-
Prospects for Creation of Cardioprotective and Antiarrhythmic Drugs Based on Opioid Receptor Agonists.Med Res Rev. 2016 Sep;36(5):871-923. doi: 10.1002/med.21395. Epub 2016 May 16. Med Res Rev. 2016. PMID: 27197922 Free PMC article. Review.
References
-
- Goldin AL. Voltage-gated sodium channels. In: North RA, editor. Ligand- and Voltage-Gated Ion Channels. Boca Raton: CRC Press; 1995. pp. 73–112.
-
- Noda M, Ikeda T, Suzuki H, et al. Expression of functional Na+ channels from cloned cDNA. Nature. 1986;322:826–8. - PubMed
-
- Isom LL, DeJongh KS, Patton DE, et al. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256:839–42. - PubMed
-
- Goldin AL. Molecular analysis of Na+ channel inactivation. In: Perracjia C, editor. Hanbook of Membrane Channels, vol 1. San Diego: Academic Press; 1994. pp. 121–35.
LinkOut - more resources
Full Text Sources