

Review
doi: 10.1038/nchem.297.
Enantioselective protonation
Affiliations
- PMID: 20428461
- PMCID: PMC2860147
- DOI: 10.1038/nchem.297
Item in Clipboard
Review
Enantioselective protonation
Nat Chem.
2009 Aug.
Abstract
Enantioselective protonation is a common process in biosynthetic sequences. The decarboxylase and esterase enzymes that effect this valuable transformation are able to control both the steric environment around the proton acceptor (typically an enolate) and the proton donor (typically a thiol). Recently, several chemical methods to achieve enantioselective protonation have been developed by exploiting various means of enantiocontrol in different mechanisms. These laboratory transformations have proven useful for the preparation of a number of valuable organic compounds.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

Fehr demonstrated that enols in the presence of a chiral Lewis base may be transformed into enantioenriched ketones. This indicates a possible alternative mechanism for enantioselective protonation and suggests that sometimes these transformations may be better described as enantioselective tautomerization rather than protonation. (Reduced to 45%)
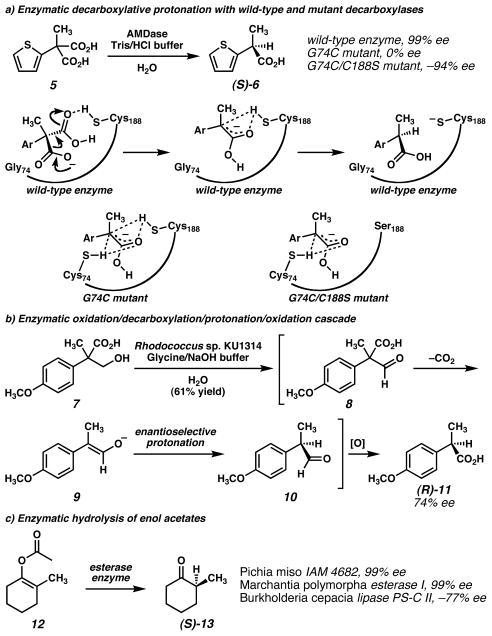
a) Ohta and coworkers, found AMDase transformed arylmalonic acids into enantioenriched arylpropionic acids. Noting the homology of AMDase to racemase enzymes led to the design of mutant enzymes that formed racemic or enantiomeric arylpropionic acids., b) Ohta and coworkers identified a Rhodococcus capable of initiating an oxidation/decarboxylation/protonation/oxidation cascade to convert tropate derivatives to enantioenriched propionic acids. c) Several esterase enzymes have been found to convert enol acetates to enantioenriched ketones.,, (Reduced to 45%)

a) Kim and coworkers synthesized several chiral alcohols (e.g., 16) to serve as chiral Brønsted acids. High e.e. products could be obtained with tetralone-derived enolates, but lower selectivity was found for substrates lacking an aryl group. b) Eames and coworkers,, employed a series of sulfonamide Brønsted acids (e.g., 18) in enantioselective protonation reactions. In some cases an external quench procedure allowed preparation of the antipode of the ketone product. c) Rouden and coworkers, found that Cincona alkaloid-derived thioureas (e.g., 21) promoted the decarboxylative protonation of malonate hemiesters. Use of diastereomeric thioureas allowed access to the opposite enantiomer of product with comparable e.e. d) Donohoe and coworkers, used amino alcohols (e.g., 23) as proton donors in dissolving metal reductions of pyrroles. In the case of diester 22, a 1:1 mixture of diastereomeric dihydropyrrole products (24) was obtained.
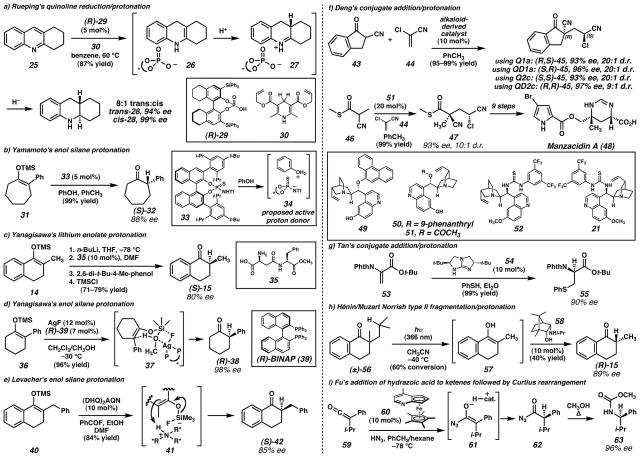
a) Rueping and coworkers found that chiral phosphoric acid catalysts coupled with Hantzch esters could effect enantioselective enamine protonation in the course of quinoline reductions. b) Cheon and Yamamoto recently disclosed the use of N-triflyl thiophosphoramide catalyst 33 for the conversion of enol silanes to enantioenriched ketones. Empirical observations suggest that the catalyst forms an oxonium ion pair (34) that serves as the active proton donor. c) Yanagisawa and coworkers used a catalytic amount simple dipeptide 35 to enantioselectively protonate lithium enolates. Small modifications to the dipeptide caused significant decreases in product e.e. d) A different system developed by Yanagisawa and coworkers, employed a Ag Lewis acid to activate methanol to serve as a proton donor in the conversion of silyl enol ethers to ketones. An aggregate transition state was proposed for the transformation. e) Levacher and coworkers reported a protocol for preparing HF in situ. The HF then interacts with a Cinchona alkaloid derived nitrogen base and subsequently activates enol silanes toward protonation by the ammonium species. f) Deng and coworkers, demonstrated that catalysts derived from quinine and quinidine were capable of catalyzing addition of prochiral nucleophiles to 2-chloroacrylonitrile (44). All four stereoisomers of the products (e.g., 45) were accessible in high yield, e.e., and diastereomeric ratio by using different catalysts. This transformation was used to carry out an enantioselective formal synthesis of the natural product manzacidin A (48). g) Tan and coworkers developed enantioselective conjugate addition/protonation sequences with heteroatom nucleophiles in the presence of guanidine catalyst 54. h) Hénin, Muzart, and coworkers, found the photochemical fragmentation of racemic α-quaternary ketone 56 generated an enol (57) that could undergo enantioselective conversion to (R)-2-methyltetralone ((R)-15) in the presence of amino alcohol catalyst 58. i) Fu and coworkers employed planar-chiral heterocycle catalyst 60 to promote addition of hydrazoic acid to ketenes (e.g., 59). Subseqeunt Curtius rearrangement generated chiral carbamate products (e.g., 63) with high e.e. (Reduced to 45%)
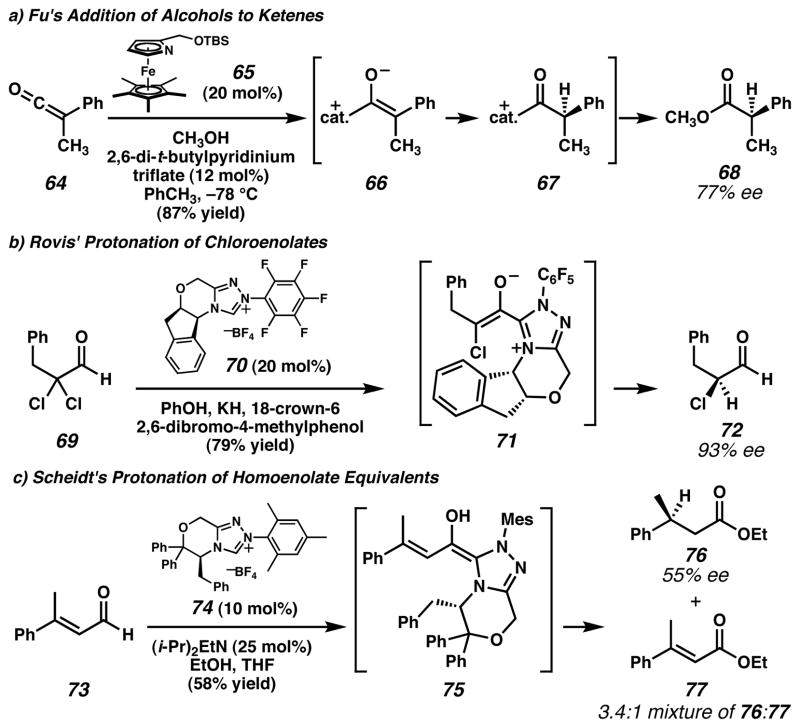
a) Fu and coworkers employed azaferrocene catalyst 65 to achieve enantioselective addition of alcohols to ketenes. Experimental observations suggest that the mechanism differs from the related reaction in Figure 4i. b) Rovis and coworkers found that triazolium salt 70 catalyzed the transformation of α,α-dichloraldehydes (e.g., 69) into synthetically useful enantioenriched mono-chloroaldehydes (e.g., 72). c) Scheidt and coworkers achieved enantioselective protonation of homoenolate equivalents with triazolium salt catalyst 74. This demonstrated that enantioselective protonation could occur at a site distant from the chiral catalyst. (Reduced to 45%)
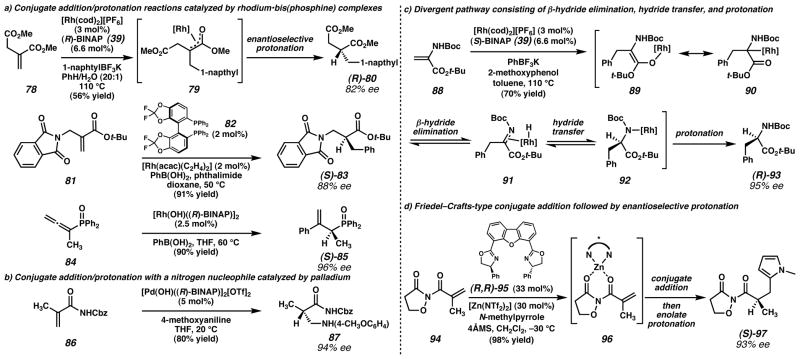
a) Several other research groups,,, have explored Rh-catalyzed conjugate addition/protonation sequences with various activated electrophiles and carbon-based nucleophiles. b) Sodeoka and coworkers found that nitrogen nucleophiles could undergo enantioselective conjugate addition/protonation in the presence of a cationic Pd catalyst. c) Darses, Genet, and coworkers, successfully synthesized α-amino acid derivatives through a conjugate addition/protonation sequence, although later mechanistic evidence suggested a different reaction mechanism than they originally postulated. d) Sibi and coworkers achieved enantioselective protonation of Zn-enolate generated by Friedel–Crafts-type addition of a pyrrole to an acrylimide. (Reduced to 45%)
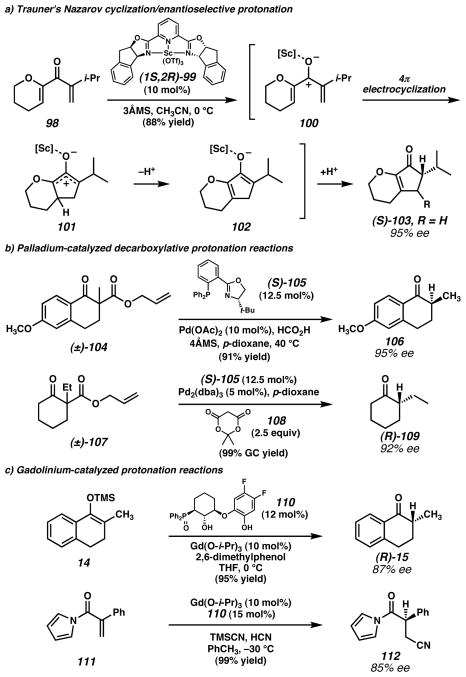
a) Trauner and coworkers developed a scandium-catalyzed cascade Nazarov cyclization/enolate protonation sequence to access cyclopentenones. b) Stoltz and coworkers, employed a chiral palladium complex to effect decarboxylative protonation reactions from allyl β-ketoester substrates (e.g., 104 and 107). Two different proton sources were effective in this transformation: formic acid in the presence of molecular sieves, and Meldrum’s acid (108). c) Kanai, Shibasaki, and coworkers found that a chiral gadolinium complex was useful for enantioselective protonation of enolates generated from either enol silanes or conjugate addition of cyanide to N-acryloyl pyrroles. (Reduced to 45%)

Sibi and coworkers, demonstrated that Mg•bis(oxazoline) complexes effectively promoted radical conjugate addition to enoates followed by enantioselective quenching of the radical intermediate by H-atom transfer. High e.e. was observed with stoichiometric amounts of Lewis acid, but in some cases substoichiometric amounts of Lewis acid led to significantly lower product e.e.
References
-
- Fehr C. Enantioselective protonation of enolates and enols. Angew Chem, Int Ed Engl. 1996;35:2566–2587.
-
- Yanagisawa A, Ishihara K, Yamamoto H. Asymmetric protonations of enol derivatives. Synlett. 1997:411–420.
-
- Eames J, Weerasooriya N. Recent advances into the enantioselective protonation of protostereogenic enol derivatives. Tetrahedron: Asymmetry. 2001;12:1–24.
-
- Duhamel L, Duhamel P, Plaquevent JC. Enantioselective protonations: Fundamental insights and new concepts. Tetrahedron: Asymmetry. 2004;15:3653–3691.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
