

Nuclear gene phylogeography using PHASE: dealing with unresolved genotypes, lost alleles, and systematic bias in parameter estimation
- PMID: 20429950
- PMCID: PMC2880299
- DOI: 10.1186/1471-2148-10-118
Nuclear gene phylogeography using PHASE: dealing with unresolved genotypes, lost alleles, and systematic bias in parameter estimation
Abstract
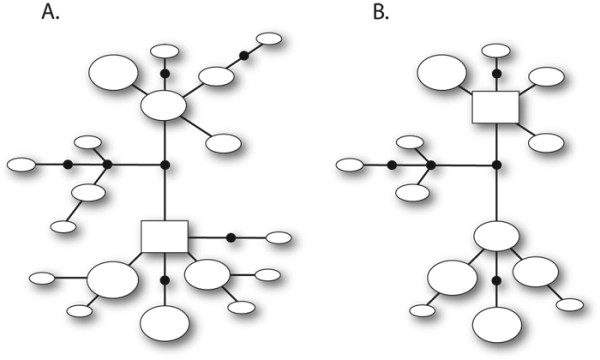
Background: A widely-used approach for screening nuclear DNA markers is to obtain sequence data and use bioinformatic algorithms to estimate which two alleles are present in heterozygous individuals. It is common practice to omit unresolved genotypes from downstream analyses, but the implications of this have not been investigated. We evaluated the haplotype reconstruction method implemented by PHASE in the context of phylogeographic applications. Empirical sequence datasets from five non-coding nuclear loci with gametic phase ascribed by molecular approaches were coupled with simulated datasets to investigate three key issues: (1) haplotype reconstruction error rates and the nature of inference errors, (2) dataset features and genotypic configurations that drive haplotype reconstruction uncertainty, and (3) impacts of omitting unresolved genotypes on levels of observed phylogenetic diversity and the accuracy of downstream phylogeographic analyses.
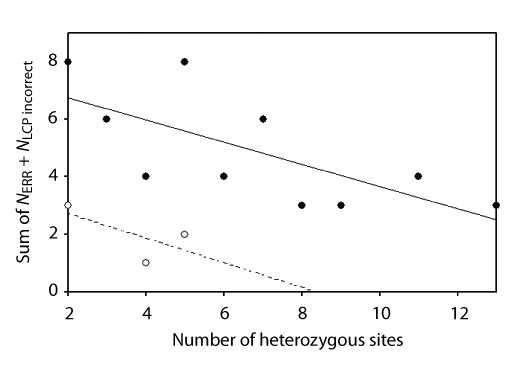
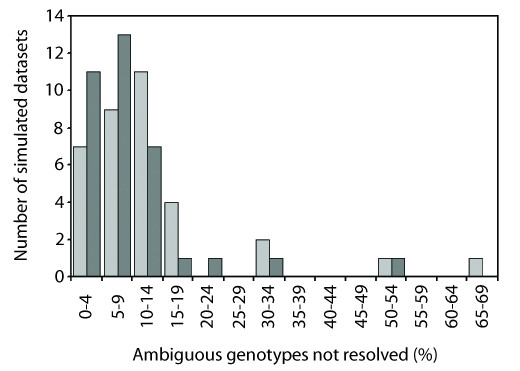
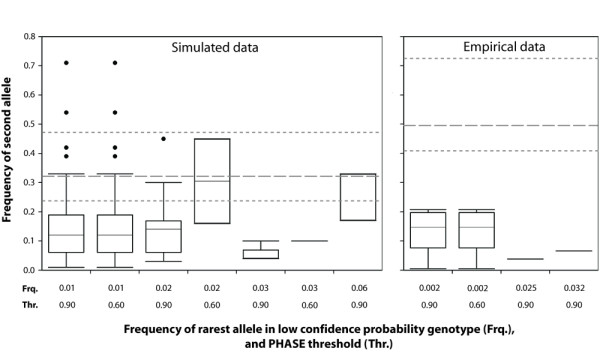
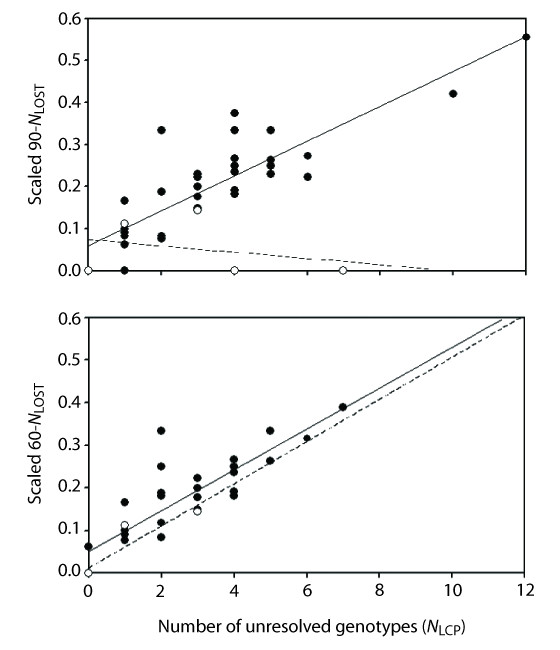
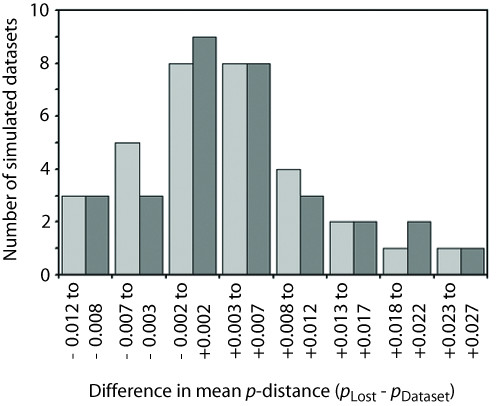
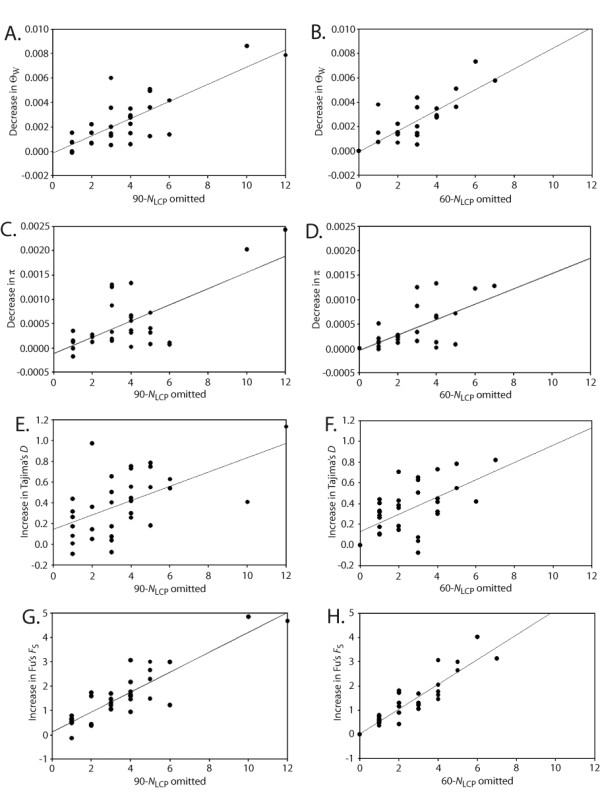
Results: We found that PHASE usually had very low false-positives (i.e., a low rate of confidently inferring haplotype pairs that were incorrect). The majority of genotypes that could not be resolved with high confidence included an allele occurring only once in a dataset, and genotypic configurations involving two low-frequency alleles were disproportionately represented in the pool of unresolved genotypes. The standard practice of omitting unresolved genotypes from downstream analyses can lead to considerable reductions in overall phylogenetic diversity that is skewed towards the loss of alleles with larger-than-average pairwise sequence divergences, and in turn, this causes systematic bias in estimates of important population genetic parameters.
Conclusions: A combination of experimental and computational approaches for resolving phase of segregating sites in phylogeographic applications is essential. We outline practical approaches to mitigating potential impacts of computational haplotype reconstruction on phylogeographic inferences. With targeted application of laboratory procedures that enable unambiguous phase determination via physical isolation of alleles from diploid PCR products, relatively little investment of time and effort is needed to overcome the observed biases.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
