

SATCHMO-JS: a webserver for simultaneous protein multiple sequence alignment and phylogenetic tree construction
- PMID: 20430824
- PMCID: PMC2896197
- DOI: 10.1093/nar/gkq298
SATCHMO-JS: a webserver for simultaneous protein multiple sequence alignment and phylogenetic tree construction
Abstract
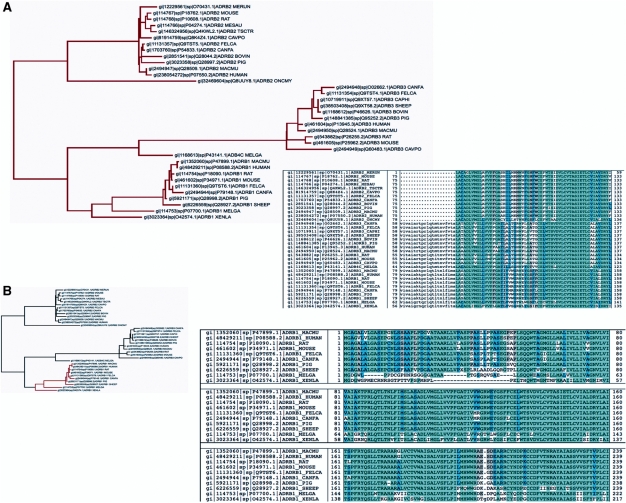
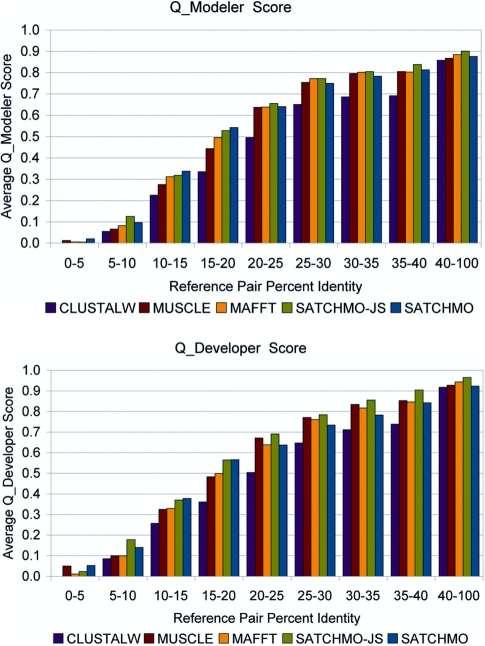
We present the jump-start simultaneous alignment and tree construction using hidden Markov models (SATCHMO-JS) web server for simultaneous estimation of protein multiple sequence alignments (MSAs) and phylogenetic trees. The server takes as input a set of sequences in FASTA format, and outputs a phylogenetic tree and MSA; these can be viewed online or downloaded from the website. SATCHMO-JS is an extension of the SATCHMO algorithm, and employs a divide-and-conquer strategy to jump-start SATCHMO at a higher point in the phylogenetic tree, reducing the computational complexity of the progressive all-versus-all HMM-HMM scoring and alignment. Results on a benchmark dataset of 983 structurally aligned pairs from the PREFAB benchmark dataset show that SATCHMO-JS provides a statistically significant improvement in alignment accuracy over MUSCLE, Multiple Alignment using Fast Fourier Transform (MAFFT), ClustalW and the original SATCHMO algorithm. The SATCHMO-JS webserver is available at http://phylogenomics.berkeley.edu/satchmo-js. The datasets used in these experiments are available for download at http://phylogenomics.berkeley.edu/satchmo-js/supplementary/.
Figures
References
-
- Sjölander K. Phylogenomic inference of protein molecular function: advances and challenges. Bioinformatics. 2004;20:170–179. - PubMed
-
- Dobzhansky CT. Nothing in biology makes sense except in the light of evolution. Am. Biol. Teach. 1973;35:125–129.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
