

Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-α-dependent manner
- PMID: 20431601
- PMCID: PMC3104849
- DOI: 10.1038/cdd.2010.44
Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-α-dependent manner
Abstract
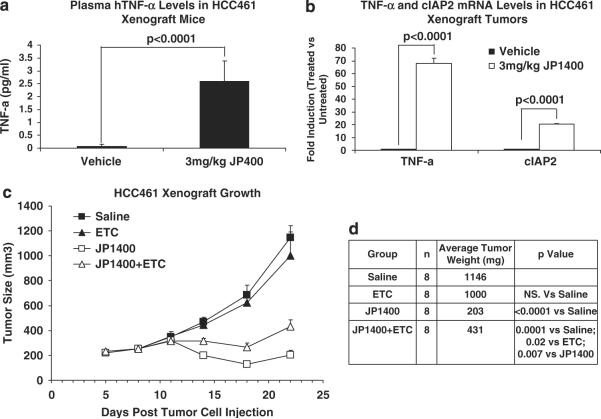
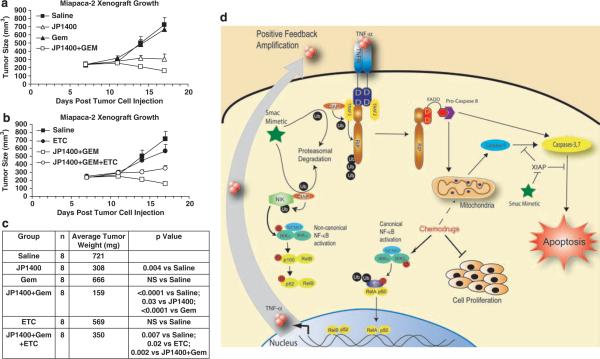
Second mitochondria-derived activator of caspase (Smac) is a mitochondrial protein released into the cytosol during apoptosis. Smac mimetics have recently been touted as a novel therapeutic to induce apoptosis in cancer cells. The ability of Smac mimetics to induce apoptosis in vitro has been shown to be dependent upon both XIAP neutralization and cancer cell autocrine tumor necrosis factor-α (TNF-α) production. In this study we provide new evidence for the utility of Smac mimetics in combination with conventional chemotherapy agents to exacerbate caspase activation and induce cancer cell death. Furthermore, we find that the combination effect is because of a multifaceted mechanism involving both inhibition of cell proliferation by the chemotherapy agents and an enhanced autocrine TNF-α feedback loop by the Smac mimetic/chemotherapy agent combination. Surprisingly, although genotoxic agents typically induce apoptosis through the mitochondrial intrinsic pathway, we show that this synergism is mediated through a TNF-α/RIP1-dependent pathway, leading to activation of the extrinsic apoptotic pathway. Finally, we report that autocrine TNF-α contributes to Smac mimetic-induced tumor regression as a single agent or in combination with chemotherapeutics in xenograft mouse models. Collectively, we provide mechanistic and applicable data to support translational studies in the use of a Smac mimetic/chemotherapy antineoplasm modality.
Figures
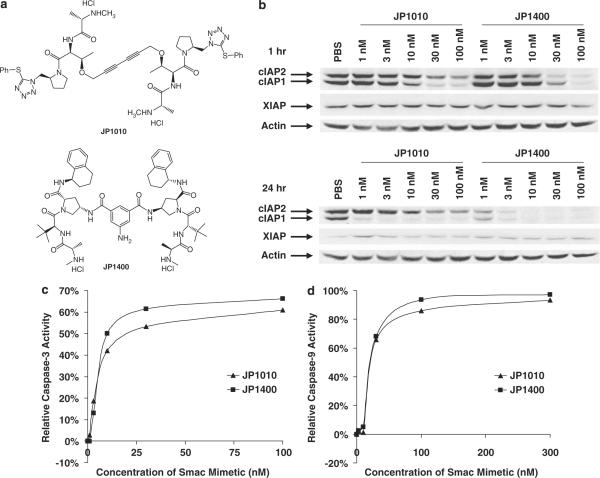
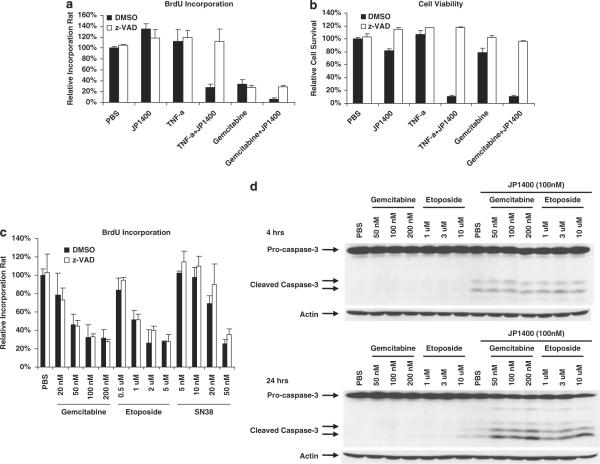
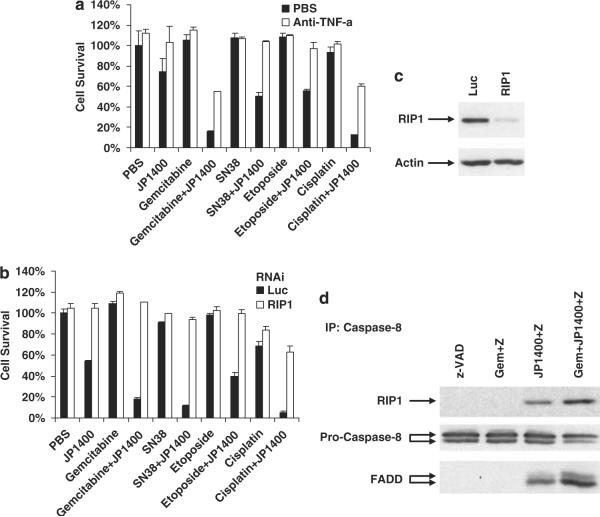
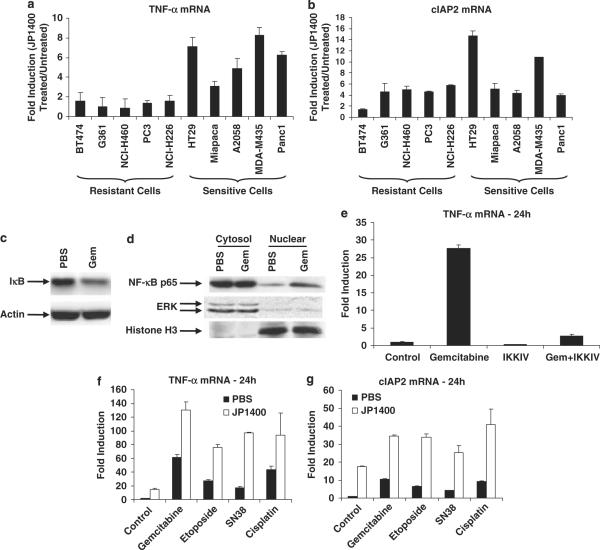
References
-
- Gorlick R, Bertino J. Drug Resistance in colon cancer. Semin Oncol. 1999;26:606–611. - PubMed
-
- Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–64. - PubMed
-
- Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–885. - PubMed
-
- Los M, Burek CJ, Stroh C, Benedyk K, Hug H, Mackiewicz A. Anticancer drugs of tomorrow: apoptotic pathways as targets for drug design. Drug Discov Today. 2003;8:67–77. - PubMed
-
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
