

MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria
- PMID: 20436477
- PMCID: PMC4070879
- DOI: 10.1038/ncb2057
MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria
Abstract
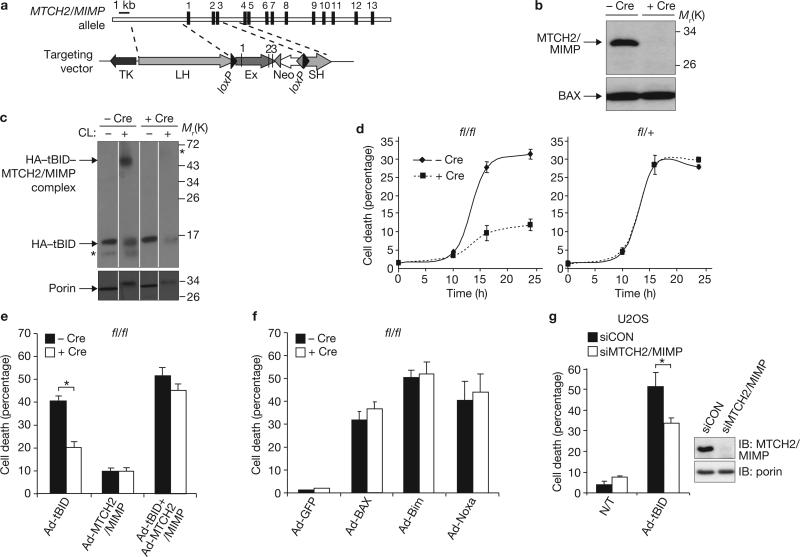
The BH3-only BID protein (BH3-interacting domain death agonist) has a critical function in the death-receptor pathway in the liver by triggering mitochondrial outer membrane permeabilization (MOMP). Here we show that MTCH2/MIMP (mitochondrial carrier homologue 2/Met-induced mitochondrial protein), a novel truncated BID (tBID)-interacting protein, is a surface-exposed outer mitochondrial membrane protein that facilitates the recruitment of tBID to mitochondria. Knockout of MTCH2/MIMP in embryonic stem cells and in mouse embryonic fibroblasts hinders the recruitment of tBID to mitochondria, the activation of Bax/Bak, MOMP, and apoptosis. Moreover, conditional knockout of MTCH2/MIMP in the liver decreases the sensitivity of mice to Fas-induced hepatocellular apoptosis and prevents the recruitment of tBID to liver mitochondria both in vivo and in vitro. In contrast, MTCH2/MIMP deletion had no effect on apoptosis induced by other pro-apoptotic Bcl-2 family members and no detectable effect on the outer membrane lipid composition. These loss-of-function models indicate that MTCH2/MIMP has a critical function in liver apoptosis by regulating the recruitment of tBID to mitochondria.
Figures






References
-
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. - PubMed
-
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature Rev. Mol. Cell Biol. 2008;9:47–59. - PubMed
-
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. - PubMed
-
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007;87:99–163. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
