

Developmental and degenerative features in a complicated spastic paraplegia
- PMID: 20437587
- PMCID: PMC3027847
- DOI: 10.1002/ana.21923
Developmental and degenerative features in a complicated spastic paraplegia
Abstract
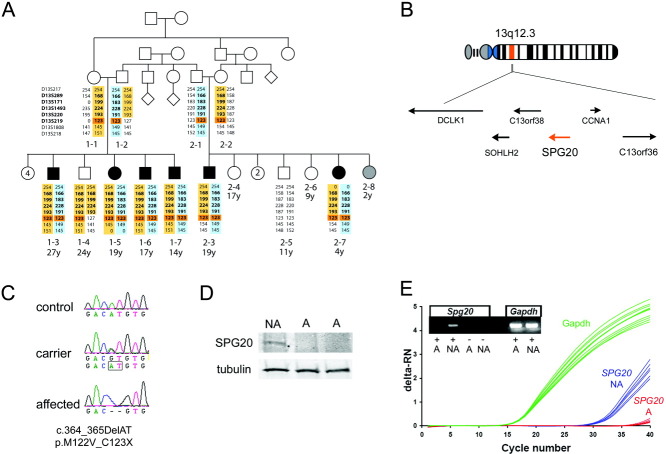
Objective: We sought to explore the genetic and molecular causes of Troyer syndrome, one of several complicated hereditary spastic paraplegias (HSPs). Troyer syndrome had been thought to be restricted to the Amish; however, we identified 2 Omani families with HSP, short stature, dysarthria and developmental delay-core features of Troyer syndrome-and a novel mutation in the SPG20 gene, which is also mutated in the Amish. In addition, we analyzed SPG20 expression throughout development to infer how disruption of this gene might generate the constellation of developmental and degenerative Troyer syndrome phenotypes.
Methods: Clinical characterization of 2 non-Amish families with Troyer syndrome was followed by linkage and sequencing analysis. Quantitative polymerase chain reaction and in situ hybridization analysis of SPG20 expression were carried out in embryonic and adult human and mouse tissue.
Results: Two Omani families carrying a novel SPG20 mutation displayed clinical features remarkably similar to the Amish patients with Troyer syndrome. SPG20 mRNA is expressed broadly but at low relative levels in the adult brain; however, it is robustly and specifically expressed in the limbs, face, and brain during early morphogenesis.
Interpretation: Null mutations in SPG20 cause Troyer syndrome, a specific clinical entity with developmental and degenerative features. Maximal expression of SPG20 in the limb buds and forebrain during embryogenesis may explain the developmental origin of the skeletal and cognitive defects observed in this disorder.
Figures



References
-
- Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. - PubMed
-
- Depienne C, Stevanin G, Brice A, Durr A. Hereditary spastic paraplegias: an update. Curr Opin Neurol. 2007;20:674–680. - PubMed
-
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. - PubMed
-
- Cross HE, McKusick VA. The Troyer syndrome. A recessive form of spastic paraplegia with distal muscle wasting. Arch Neurol. 1967;16:473–485. - PubMed
-
- Proukakis C, Cross H, Patel H, et al. Troyer syndrome revisited. A clinical and radiological study of a complicated hereditary spastic paraplegia. J Neurol. 2004;251:1105–1110. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
