

A conformal Bayesian network for classification of Mycobacterium tuberculosis complex lineages
- PMID: 20438651
- PMCID: PMC2863063
- DOI: 10.1186/1471-2105-11-S3-S4
A conformal Bayesian network for classification of Mycobacterium tuberculosis complex lineages
Abstract
Background: We present a novel conformal Bayesian network (CBN) to classify strains of Mycobacterium tuberculosis Complex (MTBC) into six major genetic lineages based on two high-throuput biomarkers: mycobacterial interspersed repetitive units (MIRU) and spacer oligonucleotide typing (spoligotyping). MTBC is the causative agent of tuberculosis (TB), which remains one of the leading causes of disease and morbidity world-wide. DNA fingerprinting methods such as MIRU and spoligotyping are key components in the control and tracking of modern TB.
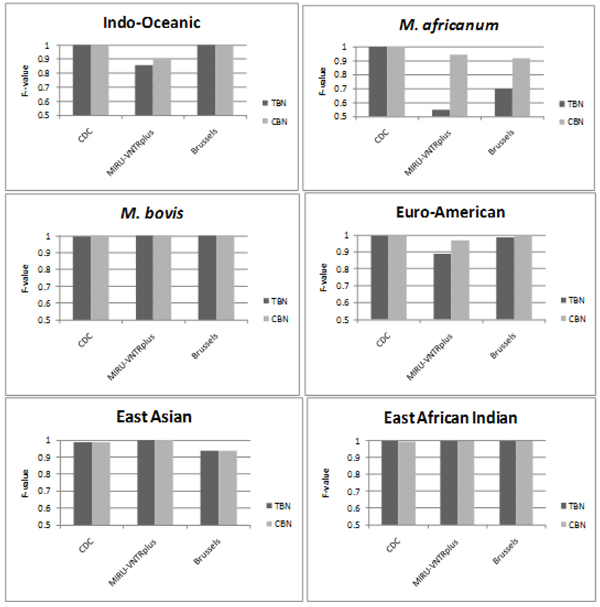
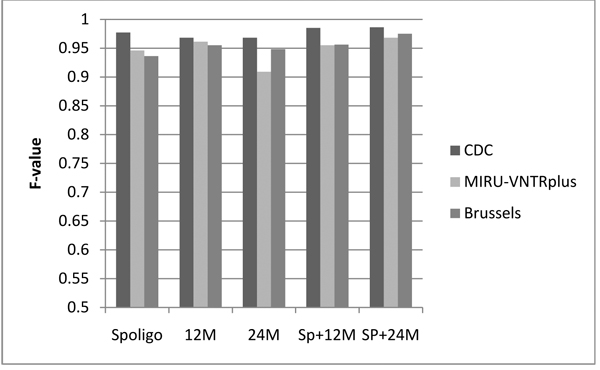
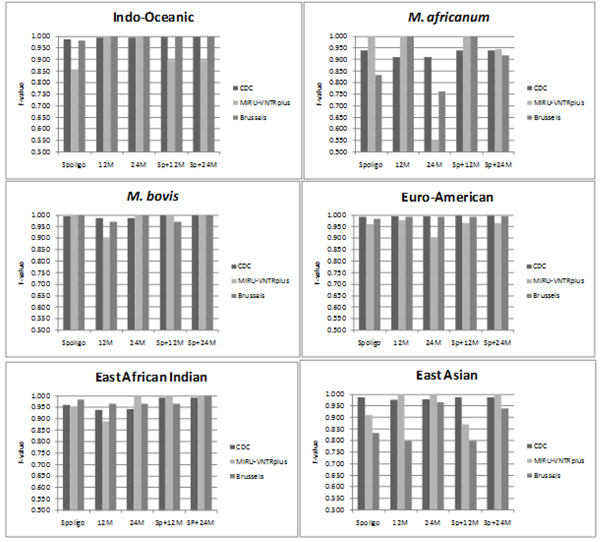
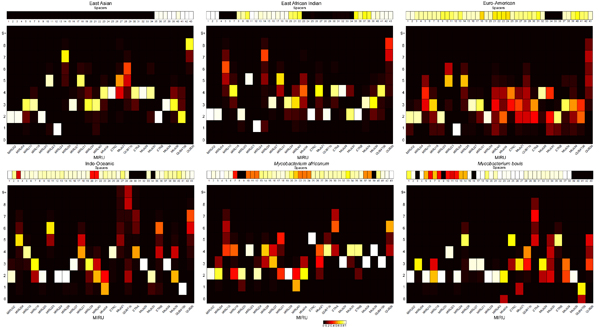
Results: CBN is designed to exploit background knowledge about MTBC biomarkers. It can be trained on large historical TB databases of various subsets of MTBC biomarkers. During TB control efforts not all biomarkers may be available. So, CBN is designed to predict the major lineage of isolates genotyped by any combination of the PCR-based typing methods: spoligotyping and MIRU typing. CBN achieves high accuracy on three large MTBC collections consisting of over 34,737 isolates genotyped by different combinations of spoligotypes, 12 loci of MIRU, and 24 loci of MIRU. CBN captures distinct MIRU and spoligotype signatures associated with each lineage, explaining its excellent performance. Visualization of MIRU and spoligotype signatures yields insight into both how the model works and the genetic diversity of MTBC.
Conclusions: CBN conforms to the available PCR-based biological markers and achieves high performance in identifying major lineages of MTBC. The method can be readily extended as new biomarkers are introduced for TB tracking and control. An online tool (http://www.cs.rpi.edu/~bennek/tbinsight/tblineage) makes the CBN model available for TB control and research efforts.
Figures
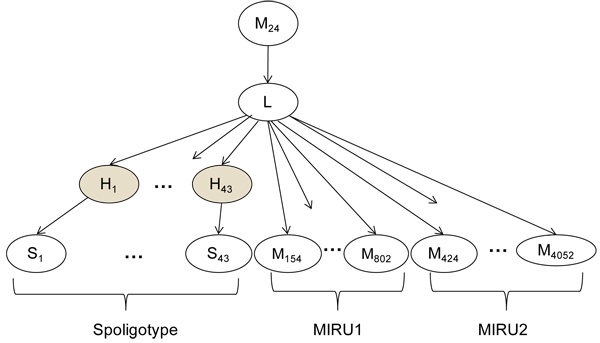
 capture the fact that spacers are lost and almost never gained. The shaded nodes refer to hidden variables.
capture the fact that spacers are lost and almost never gained. The shaded nodes refer to hidden variables.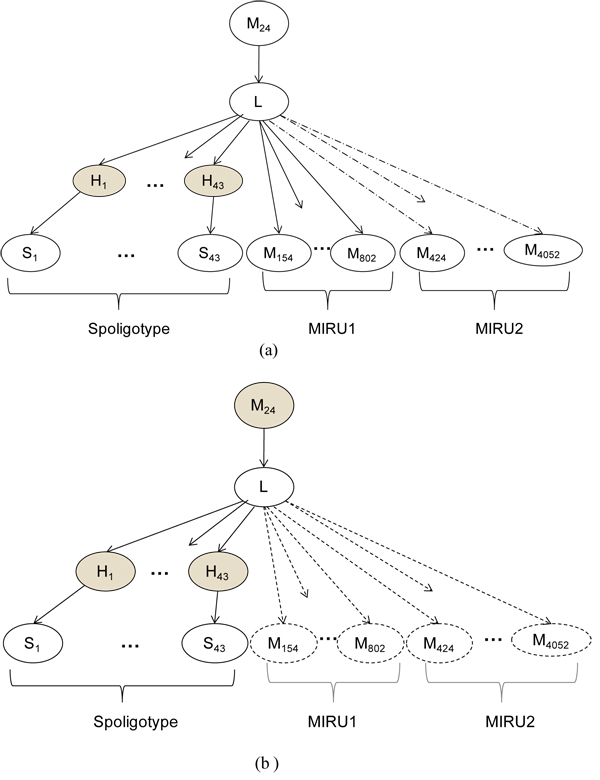
References
-
- Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rusch-Gerdes S, Willery E, Savine E, de Haas P, van Deutekom H, Roring S. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2006;44(12):4498–4510. doi: 10.1128/JCM.01392-06. - DOI - PMC - PubMed
-
- Ferdinand S, Valetudie G, Sola C, Rastogi N. Data mining of Mycobacterium tuberculosis complex genotyping results using mycobacterial interspersed repetitive units validates the clonal structure of spoligotyping-defined families. Res Microbiol. 2004;155(8):647–654. doi: 10.1016/j.resmic.2004.04.013. - DOI - PubMed
-
- Filliol I, Driscoll JR, van Soolingen D, Kreiswirth BN, Kremer K, Valetudie G, Anh DD, Barlow R, Banerjee D, Bifani PJ. Snapshot of moving and expanding clones of Mycobacterium tuberculosis and their global distribution assessed by spoligotyping in an international study. J Clin Microbiol. 2003;41(5):1963–1970. doi: 10.1128/JCM.41.5.1963-1970.2003. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
