

Extracellular signal-regulated kinase promotes Rho-dependent focal adhesion formation by suppressing p190A RhoGAP
- PMID: 20439493
- PMCID: PMC2897570
- DOI: 10.1128/MCB.01178-09
Extracellular signal-regulated kinase promotes Rho-dependent focal adhesion formation by suppressing p190A RhoGAP
Abstract
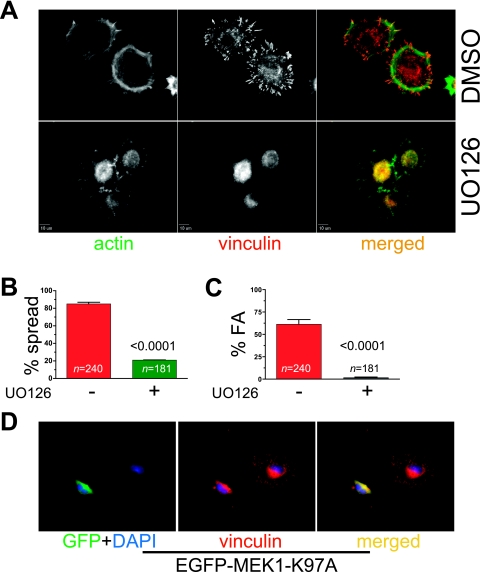
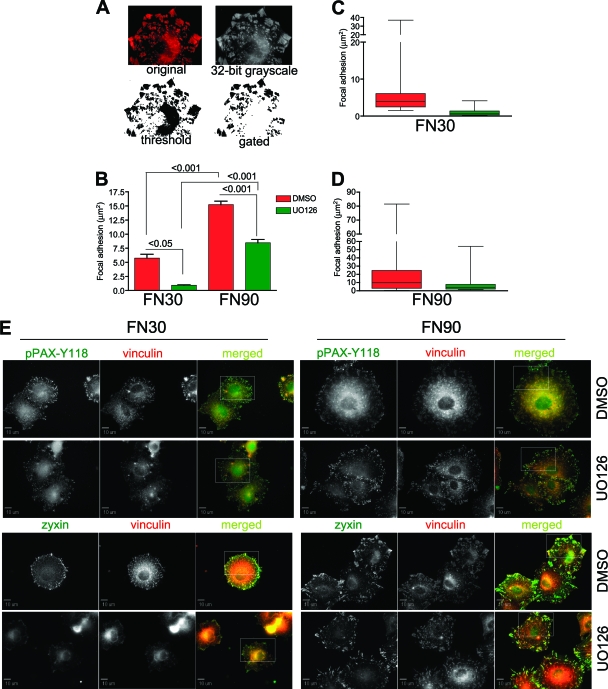
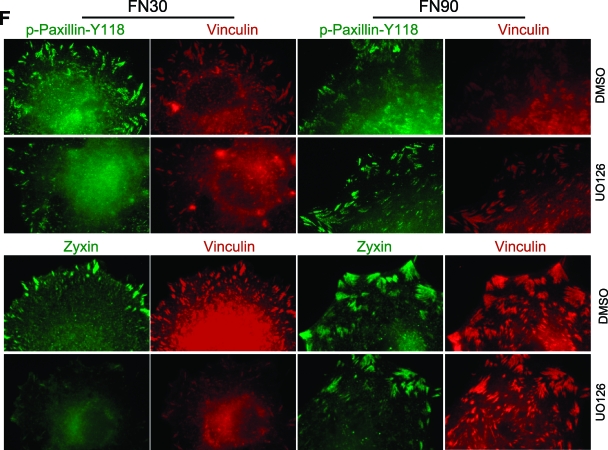
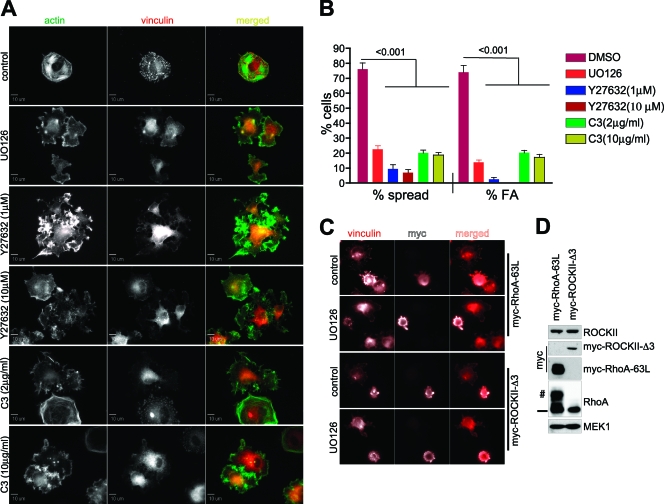
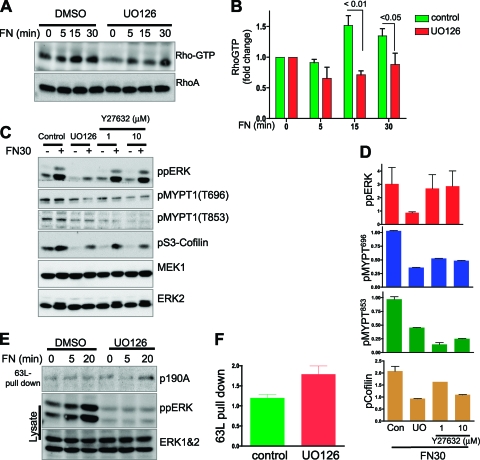
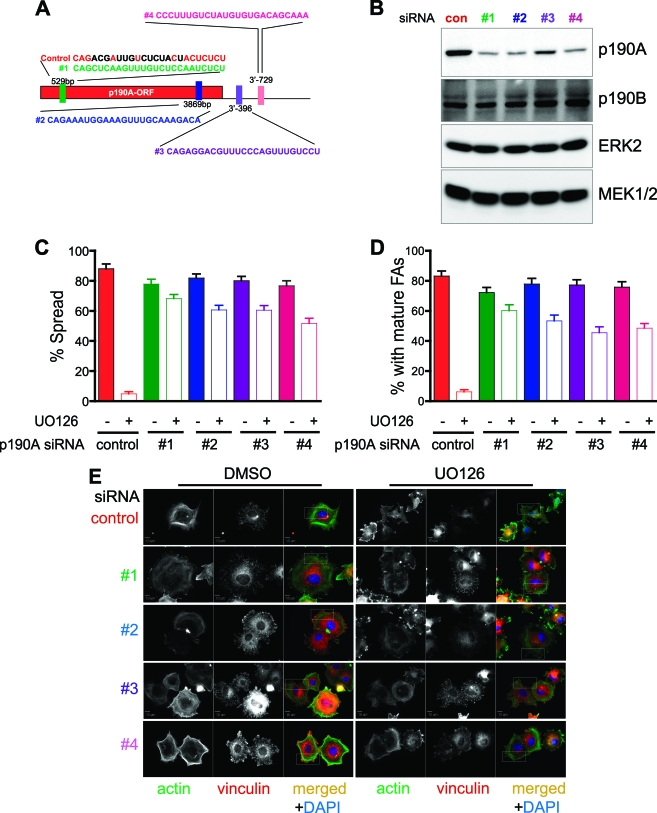
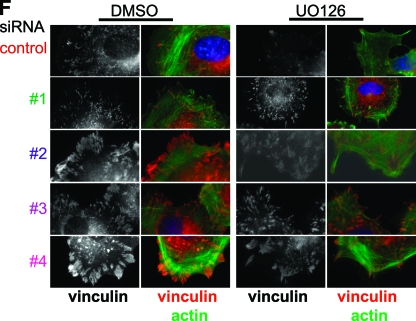
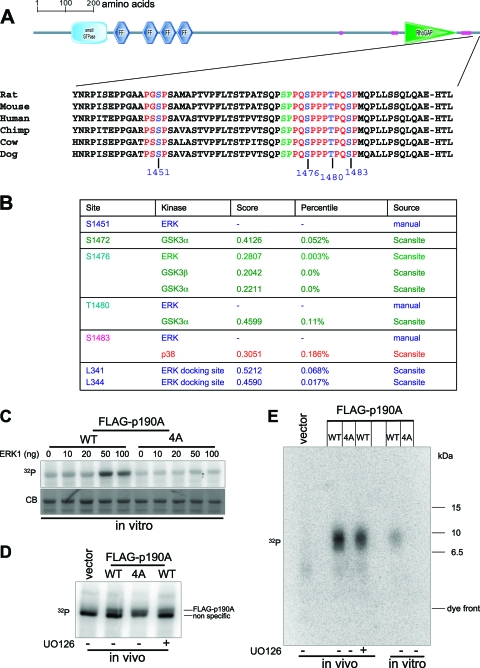
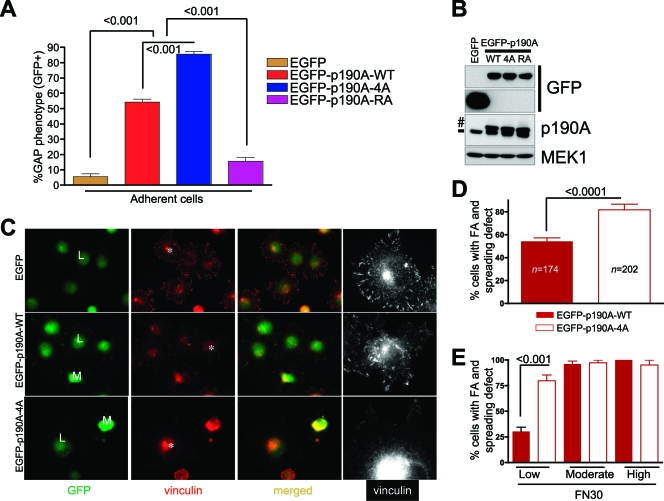
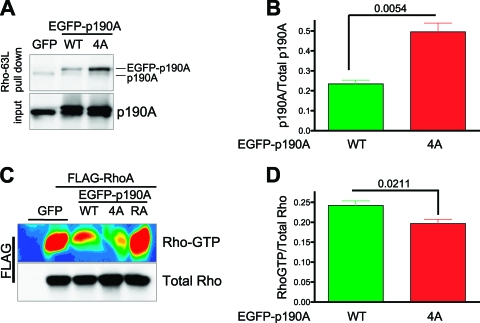
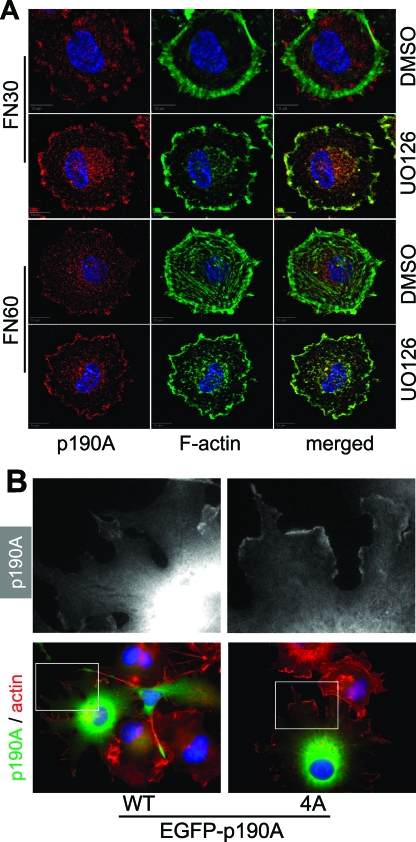
Cell migration is critical for normal development and for pathological processes including cancer cell metastasis. Dynamic remodeling of focal adhesions and the actin cytoskeleton are crucial determinants of cell motility. The Rho family and the mitogen-activated protein kinase (MAPK) module consisting of MEK-extracellular signal-regulated kinase (ERK) are important regulators of these processes, but mechanisms for the integration of these signals during spreading and motility are incompletely understood. Here we show that ERK activity is required for fibronectin-stimulated Rho-GTP loading, Rho-kinase function, and the maturation of focal adhesions in spreading cells. We identify p190A RhoGAP as a major target for ERK signaling in adhesion assembly and identify roles for ERK phosphorylation of the C terminus in p190A localization and activity. These observations reveal a novel role for ERK signaling in adhesion assembly in addition to its established role in adhesion disassembly.
Figures
Similar articles
-
The MEK1 scaffolding protein MP1 regulates cell spreading by integrating PAK1 and Rho signals.Mol Cell Biol. 2005 Jun;25(12):5119-33. doi: 10.1128/MCB.25.12.5119-5133.2005. Mol Cell Biol. 2005. PMID: 15923628 Free PMC article.
-
Rho-kinase contributes to sustained RhoA activation through phosphorylation of p190A RhoGAP.J Biol Chem. 2009 Feb 20;284(8):5067-76. doi: 10.1074/jbc.M806853200. Epub 2008 Dec 22. J Biol Chem. 2009. PMID: 19103606
-
B-RAF regulation of Rnd3 participates in actin cytoskeletal and focal adhesion organization.Mol Biol Cell. 2008 Feb;19(2):498-508. doi: 10.1091/mbc.e07-09-0895. Epub 2007 Nov 28. Mol Biol Cell. 2008. PMID: 18045987 Free PMC article.
-
Rho-associated kinase-dependent contraction of stress fibres and the organization of focal adhesions.J R Soc Interface. 2011 Mar 6;8(56):305-11. doi: 10.1098/rsif.2010.0419. Epub 2010 Sep 8. J R Soc Interface. 2011. PMID: 20826475 Free PMC article. Review.
-
Scaffold mediated regulation of MAPK signaling and cytoskeletal dynamics: a perspective.Cell Signal. 2007 Aug;19(8):1621-32. doi: 10.1016/j.cellsig.2007.04.012. Epub 2007 May 5. Cell Signal. 2007. PMID: 17553668 Free PMC article. Review.
Cited by
-
ZINC40099027 promotes monolayer circular defect closure by a novel pathway involving cytosolic activation of focal adhesion kinase and downstream paxillin and ERK1/2.Cell Tissue Res. 2022 Nov;390(2):261-279. doi: 10.1007/s00441-022-03674-1. Epub 2022 Aug 24. Cell Tissue Res. 2022. PMID: 36001146
-
Fyn Kinase Activity and Its Role in Neurodegenerative Disease Pathology: a Potential Universal Target?Mol Neurobiol. 2021 Nov;58(11):5986-6005. doi: 10.1007/s12035-021-02518-3. Epub 2021 Aug 25. Mol Neurobiol. 2021. PMID: 34432266 Review.
-
RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas.Cancer Discov. 2013 Apr;3(4):444-57. doi: 10.1158/2159-8290.CD-12-0388. Epub 2013 Jan 28. Cancer Discov. 2013. PMID: 23358651 Free PMC article.
-
p190A RhoGAP induces CDH1 expression and cooperates with E-cadherin to activate LATS kinases and suppress tumor cell growth.Oncogene. 2020 Aug;39(33):5570-5587. doi: 10.1038/s41388-020-1385-2. Epub 2020 Jul 8. Oncogene. 2020. PMID: 32641858 Free PMC article.
-
Fgfr-Ras-MAPK signaling is required for apical constriction via apical positioning of Rho-associated kinase during mechanosensory organ formation.Development. 2012 Sep;139(17):3130-5. doi: 10.1242/dev.082271. Epub 2012 Jul 25. Development. 2012. PMID: 22833124 Free PMC article.
References
-
- Aktories, K., and I. Just. 1995. In vitro ADP-ribosylation of Rho by bacterial ADP-ribosyltransferases. Methods Enzymol. 256:184-195. - PubMed
-
- Amano, M., K. Chihara, K. Kimura, Y. Fukata, N. Nakamura, Y. Matsuura, and K. Kaibuchi. 1997. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science 275:1308-1311. - PubMed
-
- Amano, M., H. Mukai, Y. Ono, K. Chihara, T. Matsui, Y. Hamajima, K. Okawa, A. Iwamatsu, and K. Kaibuchi. 1996. Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science 271:648-650. - PubMed
-
- Arthur, W. T., L. A. Petch, and K. Burridge. 2000. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr. Biol. 10:719-722. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous