

Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements
- PMID: 20439767
- PMCID: PMC2889096
- DOI: 10.1073/pnas.1000148107
Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements
Abstract
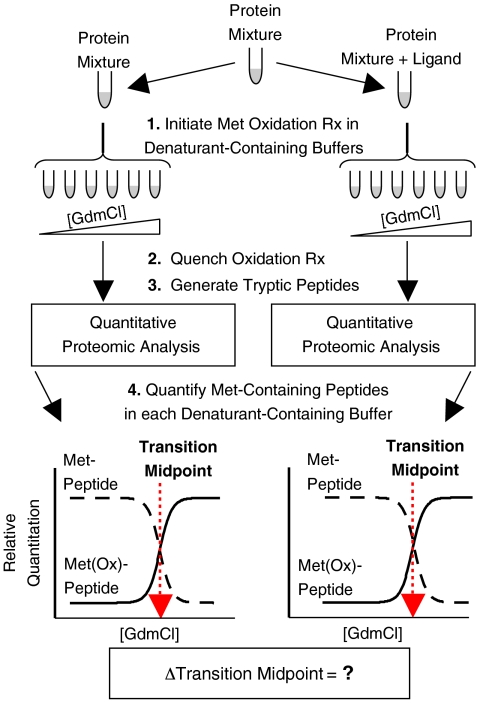
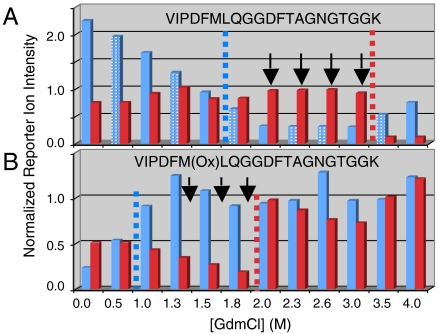
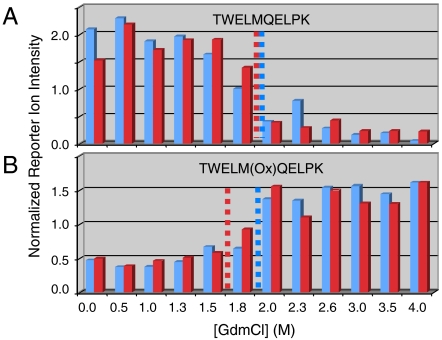
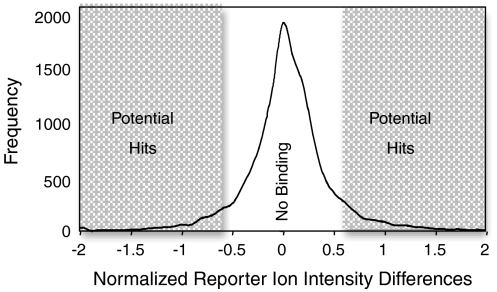
Knowledge about the protein targets of therapeutic agents is critical for understanding drug mode of action. Described here is a mass spectrometry-based proteomics method for identifying the protein target(s) of drug molecules that is potentially applicable to any drug compound. The method, which involves making thermodynamic measurements of protein-folding reactions in complex biological mixtures to detect protein-drug interactions, is demonstrated in an experiment to identify yeast protein targets of the immunosuppressive drug, cyclosporin A (CsA). Two of the ten protein targets identified in this proof of principle work were cyclophilin A and UDP-glucose-4-epimerase, both of which are known to interact with CsA, the former through a direct binding event (K(d) approximately 70 nM) and the latter through an indirect binding event. These two previously known protein targets validate the methodology and its ability to detect both the on- and off-target effects of protein-drug interactions. The other eight protein targets discovered here, which include several proteins involved in glucose metabolism, create a new framework in which to investigate the molecular basis of CsA side effects in humans.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
SILAC-pulse proteolysis: A mass spectrometry-based method for discovery and cross-validation in proteome-wide studies of ligand binding.J Am Soc Mass Spectrom. 2014 Dec;25(12):2073-83. doi: 10.1007/s13361-014-0992-y. Epub 2014 Oct 15. J Am Soc Mass Spectrom. 2014. PMID: 25315461
-
StableIsotope Labeling with Amino Acids in Cell Culture (SILAC)-based strategy for proteome-wide thermodynamic analysis of protein-ligand binding interactions.Mol Cell Proteomics. 2014 Jul;13(7):1800-13. doi: 10.1074/mcp.M113.034702. Epub 2014 Apr 16. Mol Cell Proteomics. 2014. PMID: 24741112 Free PMC article.
-
Stable isotope labeling strategy for protein-ligand binding analysis in multi-component protein mixtures.J Am Soc Mass Spectrom. 2011 Mar;22(3):418-30. doi: 10.1007/s13361-010-0060-1. Epub 2011 Feb 1. J Am Soc Mass Spectrom. 2011. PMID: 21472561 Free PMC article.
-
Profiling the Protein Targets of Unmodified Bio-Active Molecules with Drug Affinity Responsive Target Stability and Liquid Chromatography/Tandem Mass Spectrometry.Proteomics. 2020 May;20(9):e1900325. doi: 10.1002/pmic.201900325. Epub 2020 Feb 5. Proteomics. 2020. PMID: 31926115 Review.
-
Uncovering Drug Mechanism of Action by Proteome Wide- Identification of Drug-Binding Proteins.Med Chem. 2017;13(6):526-535. doi: 10.2174/1573406413666170518154724. Med Chem. 2017. PMID: 28523998 Review.
Cited by
-
Global analysis of protein folding thermodynamics for disease state characterization.J Proteome Res. 2015 May 1;14(5):2287-97. doi: 10.1021/acs.jproteome.5b00057. Epub 2015 Apr 9. J Proteome Res. 2015. PMID: 25825992 Free PMC article.
-
Machine learning of metabolite-protein interactions from model-derived metabolic phenotypes.NAR Genom Bioinform. 2024 Sep 3;6(3):lqae114. doi: 10.1093/nargab/lqae114. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 39229256 Free PMC article.
-
Advances in exploring the therapeutic potential of marine natural products.Pharmacol Res. 2019 Sep;147:104373. doi: 10.1016/j.phrs.2019.104373. Epub 2019 Jul 25. Pharmacol Res. 2019. PMID: 31351913 Free PMC article. Review.
-
An update of label-free protein target identification methods for natural active products.Theranostics. 2022 Jan 24;12(4):1829-1854. doi: 10.7150/thno.68804. eCollection 2022. Theranostics. 2022. PMID: 35198076 Free PMC article. Review.
-
Identification of direct protein targets of small molecules.ACS Chem Biol. 2011 Jan 21;6(1):34-46. doi: 10.1021/cb100294v. Epub 2010 Nov 30. ACS Chem Biol. 2011. PMID: 21077692 Free PMC article. Review.
References
-
- Fields S, Song OK. A novel genetic system to detect protein–protein interactions. Nature. 1989;340(6230):245–246. - PubMed
-
- Gavin AC, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415(6868):141–147. - PubMed
-
- Ho Y, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415(6868):180–183. - PubMed
-
- Uetz P, et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature. 2000;403(6770):623–627. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
