

Analysis of multiple ethyl methanesulfonate-mutagenized Caenorhabditis elegans strains by whole-genome sequencing
- PMID: 20439776
- PMCID: PMC2881126
- DOI: 10.1534/genetics.110.116319
Analysis of multiple ethyl methanesulfonate-mutagenized Caenorhabditis elegans strains by whole-genome sequencing
Abstract
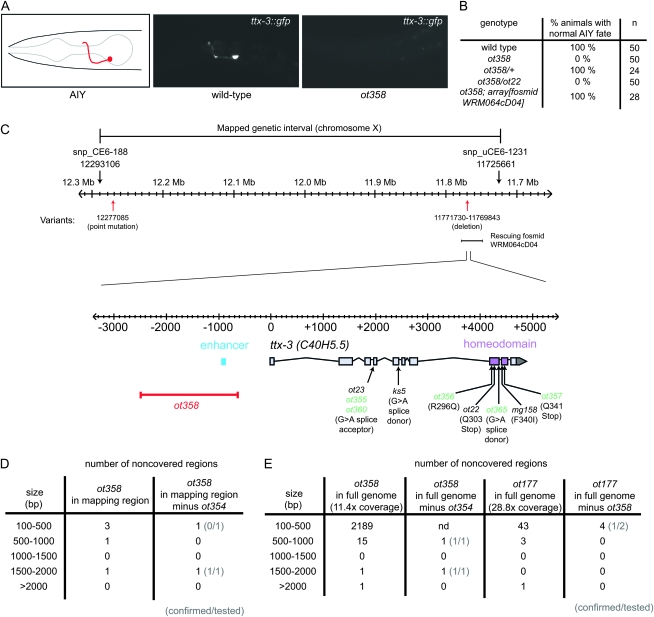
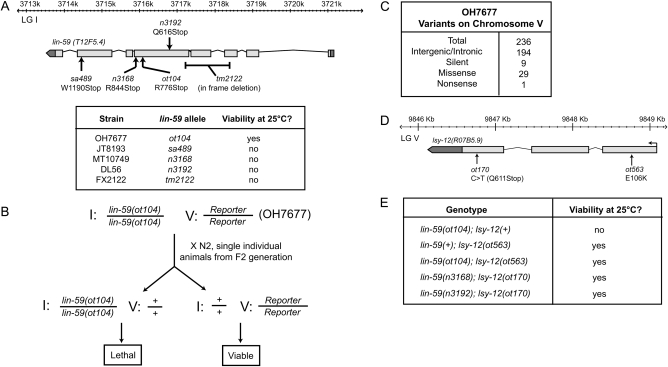
Whole-genome sequencing (WGS) of organisms displaying a specific mutant phenotype is a powerful approach to identify the genetic determinants of a plethora of biological processes. We have previously validated the feasibility of this approach by identifying a point-mutated locus responsible for a specific phenotype, observed in an ethyl methanesulfonate (EMS)-mutagenized Caenorhabditis elegans strain. Here we describe the genome-wide mutational profile of 17 EMS-mutagenized genomes as assessed with a bioinformatic pipeline, called MAQGene. Surprisingly, we find that while outcrossing mutagenized strains does reduce the total number of mutations, a striking mutational load is still observed even in outcrossed strains. Such genetic complexity has to be taken into account when establishing a causative relationship between genotype and phenotype. Even though unintentional, the 17 sequenced strains described here provide a resource of allelic variants in almost 1000 genes, including 62 premature stop codons, which represent candidate knockout alleles that will be of further use for the C. elegans community to study gene function.
Figures


References
-
- Anderson, P., 1995. Mutagenesis, pp. 31–58 in Caenorhabditis elegans—Modern Biological Analysis of an Organism, edited by H. F. Epstein and D. Shakes. Academic Press, New York/London/San Diego.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
