

The Gq/G11-mediated signaling pathway is critical for autocrine potentiation of insulin secretion in mice
- PMID: 20440069
- PMCID: PMC2877950
- DOI: 10.1172/JCI41541
The Gq/G11-mediated signaling pathway is critical for autocrine potentiation of insulin secretion in mice
Abstract
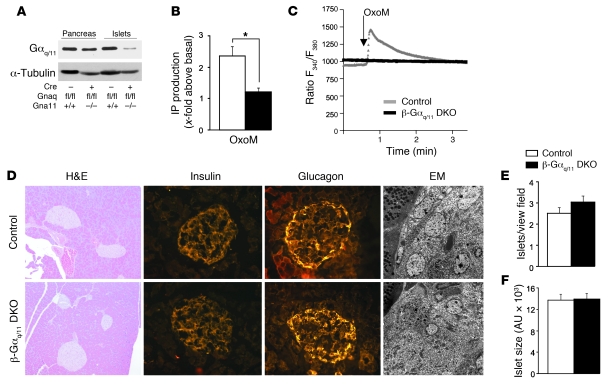
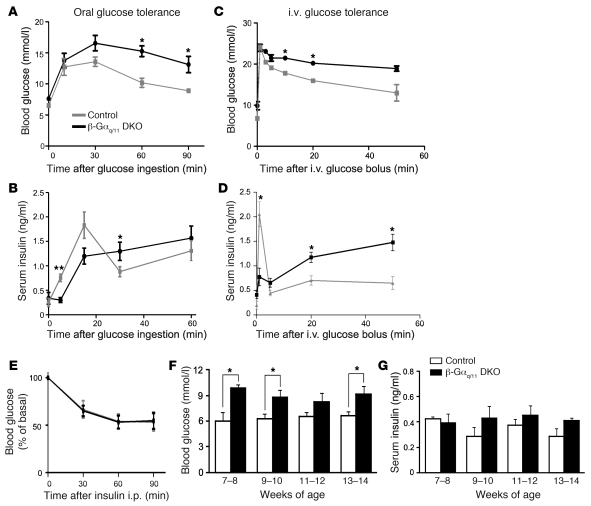
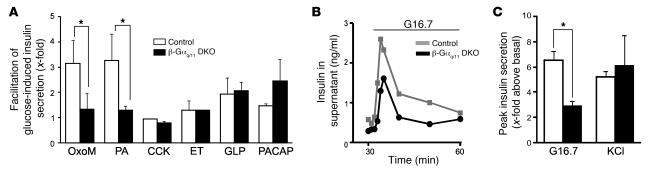
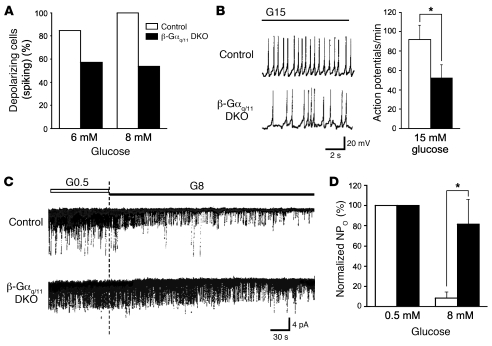
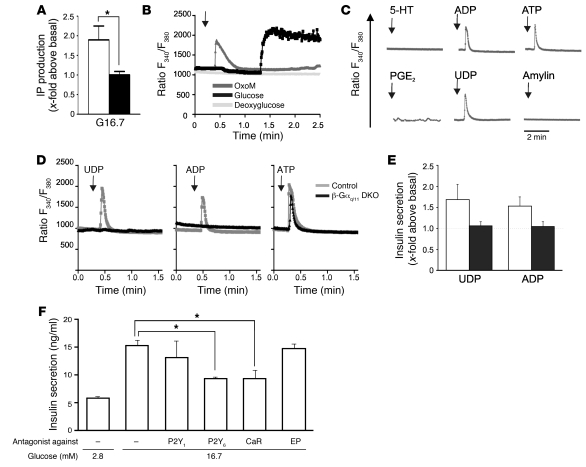
A variety of neurotransmitters, gastrointestinal hormones, and metabolic signals are known to potentiate insulin secretion through GPCRs. We show here that beta cell-specific inactivation of the genes encoding the G protein alpha-subunits Galphaq and Galpha11 resulted in impaired glucose tolerance and insulin secretion in mice. Interestingly, the defects observed in Galphaq/Galpha11-deficient beta cells were not restricted to loss of muscarinic or metabolic potentiation of insulin release; the response to glucose per se was also diminished. Electrophysiological recordings revealed that glucose-induced depolarization of isolated beta cells was impaired in the absence of Galphaq/Galpha11, and closure of KATP channels was inhibited. We provide evidence that this reduced excitability was due to a loss of beta cell-autonomous potentiation of insulin secretion through factors cosecreted with insulin. We identified as autocrine mediators involved in this process extracellular nucleotides such as uridine diphosphate acting through the Gq/G11-coupled P2Y6 receptor and extracellular calcium acting through the calcium-sensing receptor. Thus, the Gq/G11-mediated signaling pathway potentiates insulin secretion in response to glucose by integrating systemic as well as autocrine/paracrine mediators.
Figures
Similar articles
-
Gs/Gq signaling switch in β cells defines incretin effectiveness in diabetes.J Clin Invest. 2020 Dec 1;130(12):6639-6655. doi: 10.1172/JCI140046. J Clin Invest. 2020. PMID: 33196462 Free PMC article.
-
Parathyroid-specific double knockout of Gq and G11 alpha-subunits leads to a phenotype resembling germline knockout of the extracellular Ca2+ -sensing receptor.Mol Endocrinol. 2007 Jan;21(1):274-80. doi: 10.1210/me.2006-0110. Epub 2006 Sep 20. Mol Endocrinol. 2007. PMID: 16988000
-
An Acetate-Specific GPCR, FFAR2, Regulates Insulin Secretion.Mol Endocrinol. 2015 Jul;29(7):1055-66. doi: 10.1210/me.2015-1007. Epub 2015 Jun 15. Mol Endocrinol. 2015. PMID: 26075576 Free PMC article.
-
Alterations in 5-HT2A receptor signaling in male and female transgenic rats over-expressing either Gq or RGS-insensitive Gq protein.Neuropharmacology. 2006 Sep;51(3):524-35. doi: 10.1016/j.neuropharm.2006.04.012. Neuropharmacology. 2006. PMID: 16769091
-
Gq-Coupled Receptors in Autoimmunity.J Immunol Res. 2016;2016:3969023. doi: 10.1155/2016/3969023. Epub 2016 Jan 17. J Immunol Res. 2016. PMID: 26885533 Free PMC article. Review.
Cited by
-
Intracellular Ca2+ oscillations generated via the Ca2+-sensing receptor are mediated by negative feedback by PKCα at Thr888.Am J Physiol Cell Physiol. 2014 Feb 1;306(3):C298-306. doi: 10.1152/ajpcell.00194.2013. Epub 2013 Dec 11. Am J Physiol Cell Physiol. 2014. PMID: 24336654 Free PMC article.
-
Enhancement of glucose uptake in mouse skeletal muscle cells and adipocytes by P2Y6 receptor agonists.PLoS One. 2014 Dec 30;9(12):e116203. doi: 10.1371/journal.pone.0116203. eCollection 2014. PLoS One. 2014. PMID: 25549240 Free PMC article.
-
Isoprenoid Derivatives of Lysophosphatidylcholines Enhance Insulin and GLP-1 Secretion through Lipid-Binding GPCRs.Int J Mol Sci. 2021 May 27;22(11):5748. doi: 10.3390/ijms22115748. Int J Mol Sci. 2021. PMID: 34072220 Free PMC article.
-
Candidate master microRNA regulator of arsenic-induced pancreatic beta cell impairment revealed by multi-omics analysis.Arch Toxicol. 2022 Jun;96(6):1685-1699. doi: 10.1007/s00204-022-03263-9. Epub 2022 Mar 21. Arch Toxicol. 2022. PMID: 35314868 Free PMC article.
-
Minireview: Nutrient sensing by G protein-coupled receptors.Mol Endocrinol. 2013 Aug;27(8):1188-97. doi: 10.1210/me.2013-1100. Epub 2013 Jul 2. Mol Endocrinol. 2013. PMID: 23820899 Free PMC article. Review.
References
-
- Kahn CR. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes. 1994;43(8):1066–1084. - PubMed
-
- Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312(5993):446–448. - PubMed
-
- Emami S, et al. Stimulatory transducing systems in pancreatic islet cells. Ann N Y Acad Sci. 1998;865:118–131. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
