

Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate
- PMID: 20440268
- PMCID: PMC6329296
- DOI: 10.1038/onc.2010.129
Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate
Abstract
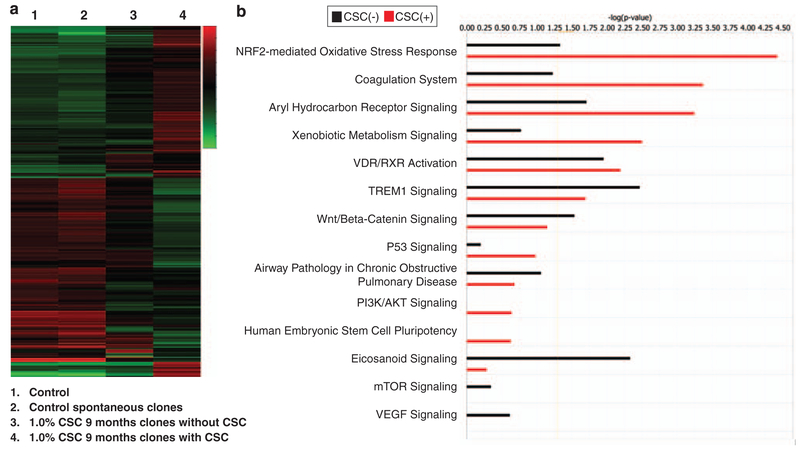
Limited information is available regarding epigenomic events mediating initiation and progression of tobacco-induced lung cancers. In this study, we established an in vitro system to examine epigenomic effects of cigarette smoke in respiratory epithelia. Normal human small airway epithelial cells and cdk-4/hTERT-immortalized human bronchial epithelial cells (HBEC) were cultured in normal media with or without cigarette smoke condensate (CSC) for up to 9 months under potentially relevant exposure conditions. Western blot analysis showed that CSC mediated dose- and time-dependent diminution of H4K16Ac and H4K20Me3, while increasing relative levels of H3K27Me3; these histone alterations coincided with decreased DNA methyltransferase 1 (DNMT1) and increased DNMT3b expression. Pyrosequencing and quantitative RT-PCR experiments revealed time-dependent hypomethylation of D4Z4, NBL2, and LINE-1 repetitive DNA sequences; up-regulation of H19, IGF2, MAGE-A1, and MAGE-A3; activation of Wnt signaling; and hypermethylation of tumor suppressor genes such as RASSF1A and RAR-beta, which are frequently silenced in human lung cancers. Array-based DNA methylation profiling identified additional novel DNA methylation targets in soft-agar clones derived from CSC-exposed HBEC; a CSC gene expression signature was also identified in these cells. Progressive genomic hypomethylation and locoregional DNA hypermethylation induced by CSC coincided with a dramatic increase in soft-agar clonogenicity. Collectively, these data indicate that cigarette smoke induces 'cancer-associated' epigenomic alterations in cultured respiratory epithelia. This in vitro model may prove useful for delineating early epigenetic mechanisms regulating gene expression during pulmonary carcinogenesis.
Conflict of interest statement
Conflict of interest
The authors declare no conflict of interest.
Figures





References
-
- Barlesi F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P et al. (2007). Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol 25: 4358–4364. - PubMed
-
- Cho HJ, Caballero OL, Gnjatic S, Andrade VC, Colleoni GW, Vettore AL et al. (2006). Physical interaction of two cancer-testis antigens, MAGE-C1 (CT7) and NY-ESO-1 (CT6). Cancer Immun 6: 12. - PubMed
-
- D’Alessio AC, Szyf M. (2006). Epigenetic tête-á-tête: the bilateral relationship between chromatin modifications and DNA methylation. Biochem Cell Biol 84: 463–476. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
