

Cardiomyocytes with disrupted CFTR function require CaMKII and Ca(2+)-activated Cl(-) channel activity to maintain contraction rate
- PMID: 20442264
- PMCID: PMC2915517
- DOI: 10.1113/jphysiol.2010.188334
Cardiomyocytes with disrupted CFTR function require CaMKII and Ca(2+)-activated Cl(-) channel activity to maintain contraction rate
Abstract
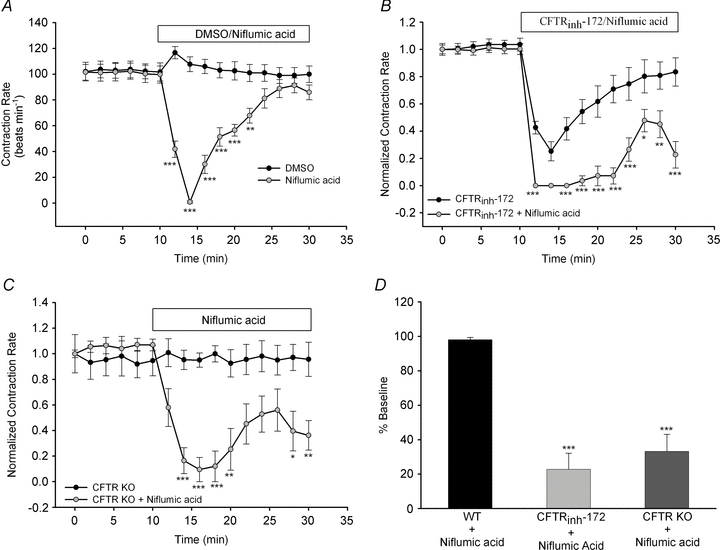
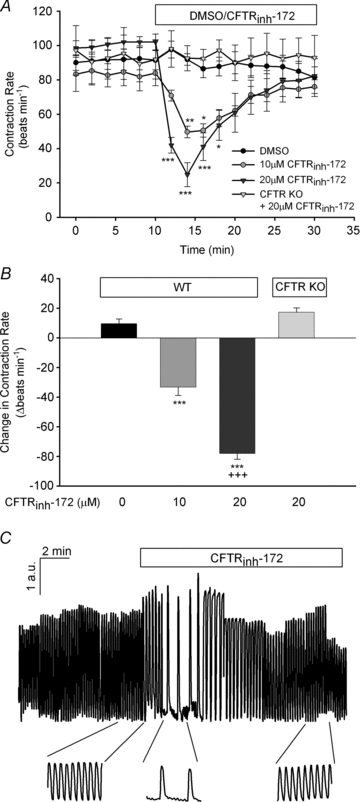
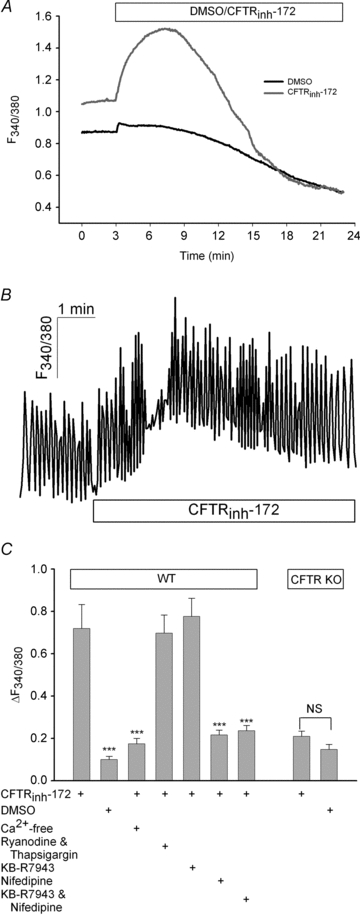
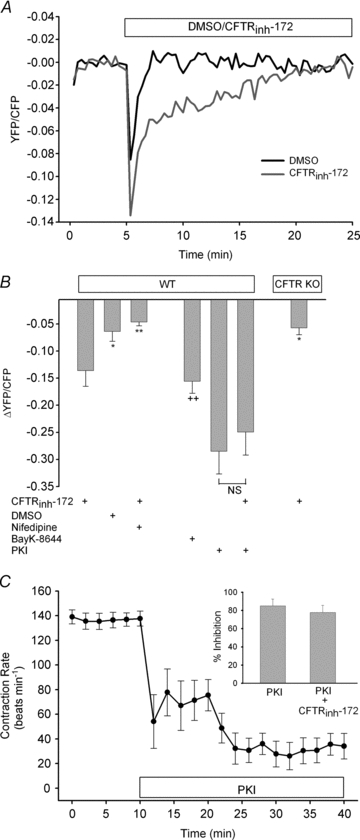
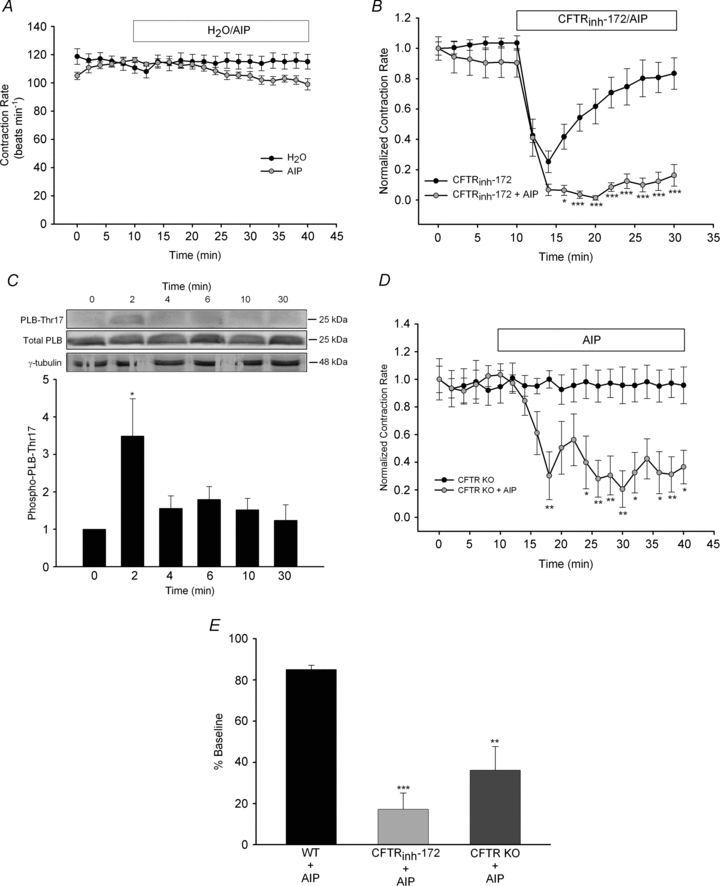
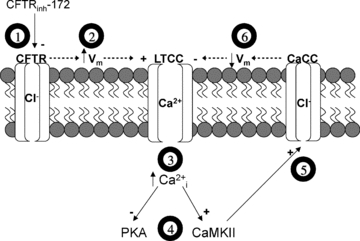
The physiological role of the cystic fibrosis transmembrane conductance regulator (CFTR) in cardiomyocytes remains unclear. Using spontaneously beating neonatal ventricular cardiomyocytes from wild-type (WT) or CFTR knockout (KO) mice, we examined the role of CFTR in the modulation of cardiomyocyte contraction rate. Contraction rates of spontaneously beating myocytes were captured by video imaging. Real-time changes in intracellular ([Ca(2+)](i)) and protein kinase A (PKA) activity were measured by fura-2 and fluorescence resonance energy transfer, respectively. Acute inhibition of CFTR in WT cardiomyocytes using the CFTR inhibitor CFTR(inh)-172 transiently inhibited the contraction rate. By contrast, cardiomyocytes from CFTR KO mice displayed normal contraction rates. Further investigation revealed that acute inhibition of CFTR activity in WT cardiomyocytes activated L-type Ca(2+) channels, leading to a transient increase of [Ca(2+)](i) and inhibition of PKA activity. Additionally, we found that contraction rate normalization following acute CFTR inhibition in WT cardiomyocytes or chronic deletion in cardiomyocytes from CFTR KO mice requires the activation of Ca(2+)/calmodulin-dependent kinase II (CaMKII) and Ca(2+)-activated Cl(-) channels (CaCC) because simultaneous addition of myristoylated-autocamtide-2-related inhibitory peptide or niflumic acid and CFTR(inh)-172 to WT cardiomyocytes or treatment of cardiomyoctes from CFTR KO mice with these agents caused sustained attenuation of contraction rates. Our results demonstrate that regulation of cardiomyocyte contraction involves CFTR. They also reveal that activation of CaMKII and CaCC compensates for loss of CFTR function. Increased dependence on CaMKII upon loss of CFTR function might leave cystic fibrosis patients at increased risk of heart dysfunction and disease.
Figures
References
-
- Ambrosi P, Chazalettes JP, Viard L, Raynaud M, Faugere G, Noirclerc M, Bernard PJ. Left ventricular involvement in mucoviscidosis after 2 years of age. Arch Fr Pediatr. 1993;50:653–656. - PubMed
-
- Anderson ME. Multiple downstream proarrhythmic targets for calmodulin kinase II: moving beyond an ion channel-centric focus. Cardiovasc Res. 2007;73:657–666. - PubMed
-
- Bers DM. Calcium cycling and signalling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. - PubMed
-
- Berul CI, Sweeten T, Vetter VL, Morad M. Lack of cystic fibrosis transmembrane regulator-type chloride current in pediatric human atrial myocytes. Life Sci. 1997;60:189–197. - PubMed
-
- Chen H, Liu LL, Ye LL, McGuckin C, Tamowski S, Scowen P, Tian H, Murray K, Hatton WJ, Duan D. Targeted inactivation of cystic fibrosis transmembrane conductance regulator chloride channel gene prevents ischemic preconditioning in isolated mouse heart. Circulation. 2004;110:700–704. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous