

Generation of cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate by CD38 for Ca2+ signaling in interleukin-8-treated lymphokine-activated killer cells
- PMID: 20442403
- PMCID: PMC2898418
- DOI: 10.1074/jbc.M109.066290
Generation of cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate by CD38 for Ca2+ signaling in interleukin-8-treated lymphokine-activated killer cells
Abstract
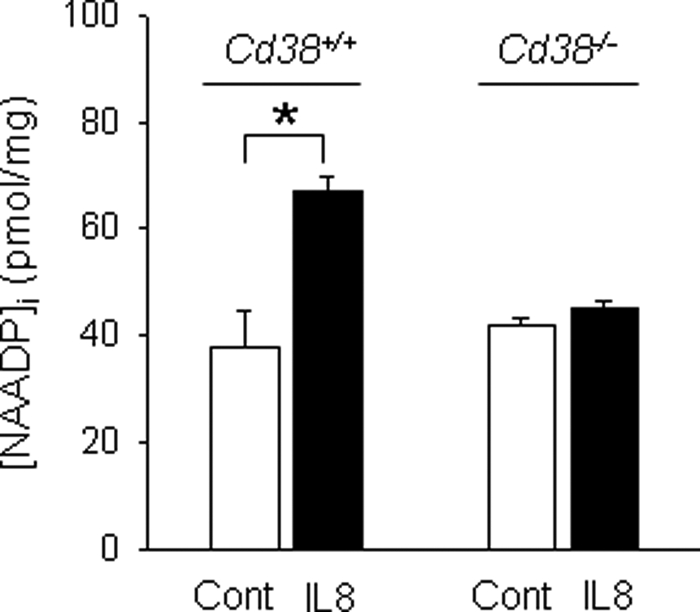
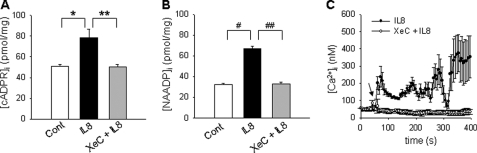
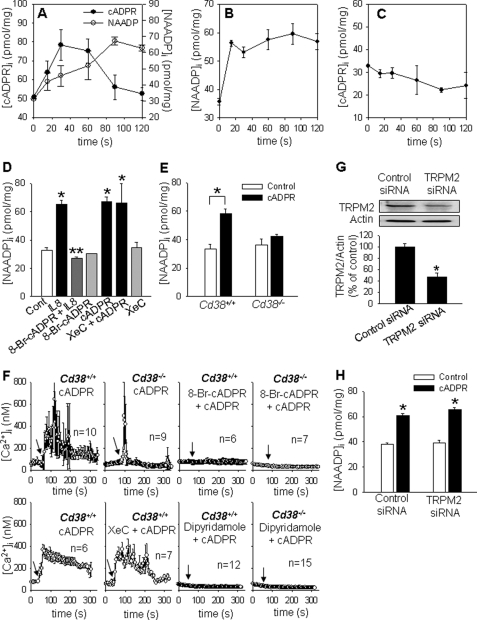
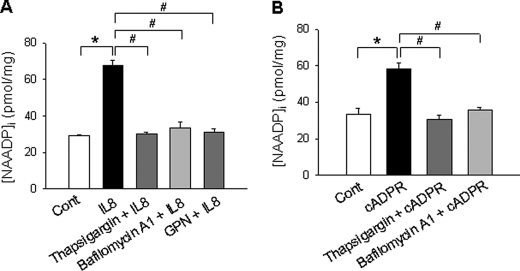
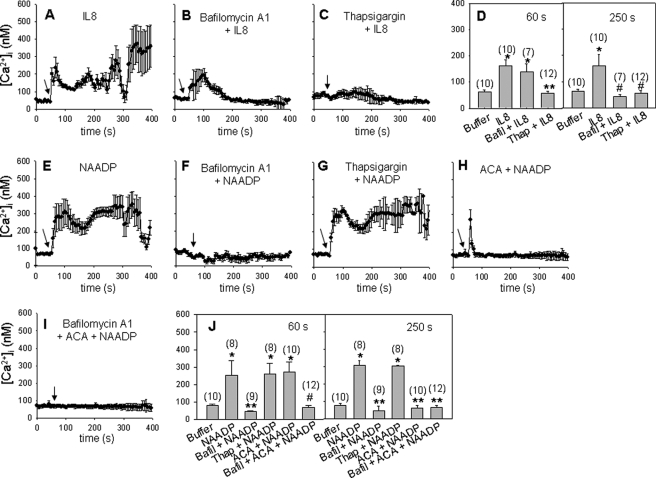
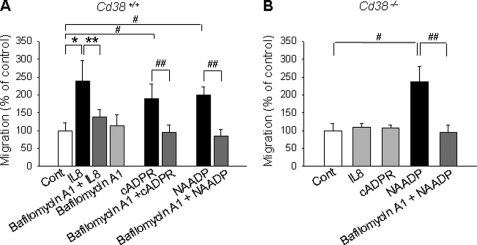
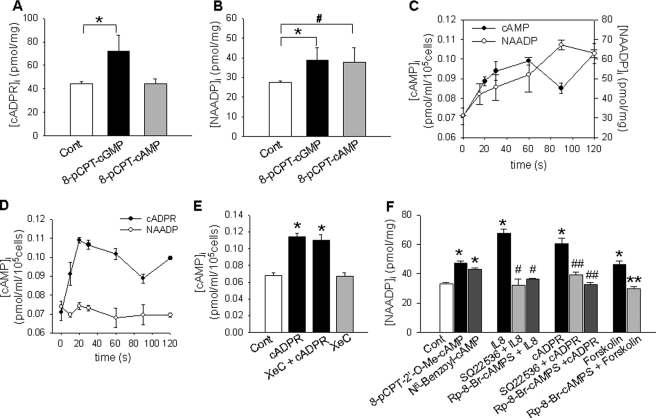
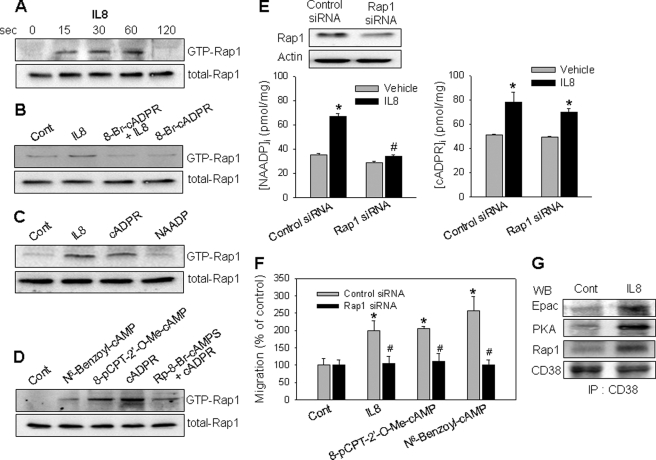
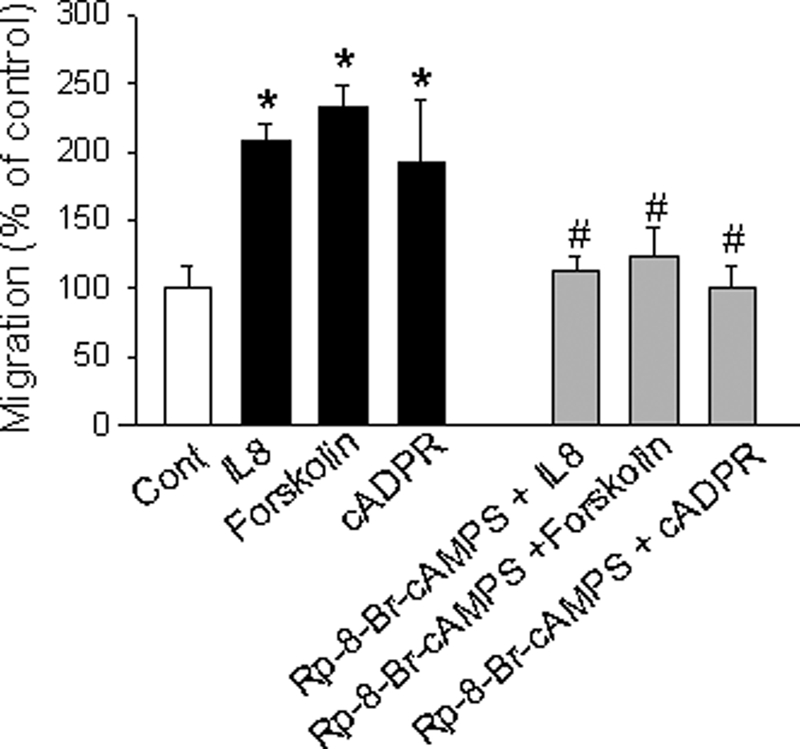
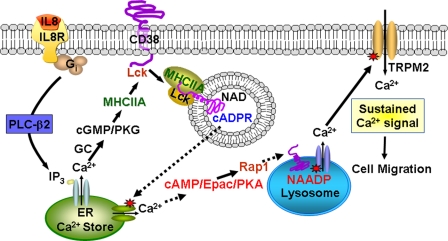
We have previously demonstrated that cyclic ADP-ribose (cADPR) is a calcium signaling messenger in interleukin 8 (IL-8)-induced lymphokine-activated killer (LAK) cells. In this study we examined the possibility that IL-8 activates CD38 to produce another messenger, nicotinic acid adenine dinucleotide phosphate (NAADP), in LAK cells, and we showed that IL-8 induced NAADP formation after cADPR production. These calcium signaling messengers were not produced when LAK cells prepared from CD38 knock-out mice were treated with IL-8, indicating that the synthesis of both NAADP and cADPR is catalyzed by CD38 in LAK cells. Application of cADPR to LAK cells induced NAADP production, whereas NAADP failed to increase intracellular cADPR levels, confirming that the production of cADPR precedes that of NAADP in IL-8-treated LAK cells. Moreover, NAADP increased intracellular Ca(2+) signaling as well as cell migration, which was completely blocked by bafilomycin A1, suggesting that NAADP is generated in lysosome-related organelles after cADPR production. IL-8 or exogenous cADPR, but not NAADP, increased intracellular cAMP levels. cGMP analog, 8-(4-chlorophenylthio)-guanosine 3',5'-cyclic monophosphate, increased both cADPR and NAADP production, whereas the cAMP analog, 8-(4-chlorophenylthio)-cAMP, increased only NAADP production, suggesting that cAMP is essential for IL-8-induced NAADP formation. Furthermore, activation of Rap1, a downstream molecule of Epac, was required for IL-8-induced NAADP formation in LAK cells. Taken together, our data suggest that IL-8-induced NAADP production is mediated by CD38 activation through the actions of cAMP/Epac/protein kinase A/Rap1 in LAK cells and that NAADP plays a key role in Ca(2+) signaling of IL-8-induced LAK cell migration.
Figures
Similar articles
-
Resolving the topological enigma in Ca2+ signaling by cyclic ADP-ribose and NAADP.J Biol Chem. 2019 Dec 27;294(52):19831-19843. doi: 10.1074/jbc.REV119.009635. Epub 2019 Oct 31. J Biol Chem. 2019. PMID: 31672920 Free PMC article. Review.
-
Activation of CD38 by interleukin-8 signaling regulates intracellular Ca2+ level and motility of lymphokine-activated killer cells.J Biol Chem. 2005 Jan 28;280(4):2888-95. doi: 10.1074/jbc.M409592200. Epub 2004 Nov 19. J Biol Chem. 2005. PMID: 15556942
-
Synthesis of the Ca2+-mobilizing messengers NAADP and cADPR by intracellular CD38 enzyme in the mouse heart: Role in β-adrenoceptor signaling.J Biol Chem. 2017 Aug 11;292(32):13243-13257. doi: 10.1074/jbc.M117.789347. Epub 2017 May 24. J Biol Chem. 2017. PMID: 28539361 Free PMC article.
-
Interleukin-8 drives CD38 to form NAADP from NADP+ and NAAD in the endolysosomes to mobilize Ca2+ and effect cell migration.FASEB J. 2020 Sep;34(9):12565-12576. doi: 10.1096/fj.202001249R. Epub 2020 Jul 27. FASEB J. 2020. PMID: 32717131
-
Mechanisms of calcium signaling by cyclic ADP-ribose and NAADP.Physiol Rev. 1997 Oct;77(4):1133-64. doi: 10.1152/physrev.1997.77.4.1133. Physiol Rev. 1997. PMID: 9354813 Review.
Cited by
-
Resolving the topological enigma in Ca2+ signaling by cyclic ADP-ribose and NAADP.J Biol Chem. 2019 Dec 27;294(52):19831-19843. doi: 10.1074/jbc.REV119.009635. Epub 2019 Oct 31. J Biol Chem. 2019. PMID: 31672920 Free PMC article. Review.
-
The role of CD38 in Fcγ receptor (FcγR)-mediated phagocytosis in murine macrophages.J Biol Chem. 2012 Apr 27;287(18):14502-14. doi: 10.1074/jbc.M111.329003. Epub 2012 Mar 6. J Biol Chem. 2012. PMID: 22396532 Free PMC article.
-
Transient receptor potential melastatin 2 channels are overexpressed in myalgic encephalomyelitis/chronic fatigue syndrome patients.J Transl Med. 2019 Dec 3;17(1):401. doi: 10.1186/s12967-019-02155-4. J Transl Med. 2019. PMID: 31796045 Free PMC article.
-
Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) and Cyclic ADP-Ribose (cADPR) Mediate Ca2+ Signaling in Cardiac Hypertrophy Induced by β-Adrenergic Stimulation.PLoS One. 2016 Mar 9;11(3):e0149125. doi: 10.1371/journal.pone.0149125. eCollection 2016. PLoS One. 2016. PMID: 26959359 Free PMC article.
-
Hippocampal mGluR1-dependent long-term potentiation requires NAADP-mediated acidic store Ca2+ signaling.Sci Signal. 2018 Nov 27;11(558):eaat9093. doi: 10.1126/scisignal.aat9093. Sci Signal. 2018. PMID: 30482851 Free PMC article.
References
-
- Lund F., Solvason N., Grimaldi J. C., Parkhouse R. M., Howard M. (1995) Immunol. Today 16, 469–473 - PubMed
-
- Mehta K., Shahid U., Malavasi F. (1996) FASEB J. 10, 1408–1417 - PubMed
-
- Kim H., Jacobson E. L., Jacobson M. K. (1993) Science 261, 1330–1333 - PubMed
-
- Howard M., Grimaldi J. C., Bazan J. F., Lund F. E., Santos-Argumedo L., Parkhouse R. M., Walseth T. F., Lee H. C. (1993) Science 262, 1056–1059 - PubMed
-
- Zocchi E., Franco L., Guida L., Benatti U., Bargellesi A., Malavasi F., Lee H. C., De Flora A. (1993) Biochem. Biophys. Res. Commun. 196, 1459–1465 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
