

Plasma pharmacokinetics and oral bioavailability of the 3,4,5,6-tetrahydrouridine (THU) prodrug, triacetyl-THU (taTHU), in mice
- PMID: 20443002
- PMCID: PMC2954253
- DOI: 10.1007/s00280-010-1337-6
Plasma pharmacokinetics and oral bioavailability of the 3,4,5,6-tetrahydrouridine (THU) prodrug, triacetyl-THU (taTHU), in mice
Abstract
Purpose: Cytidine drugs, such as gemcitabine, undergo rapid catabolism and inactivation by cytidine deaminase (CD). 3,4,5,6-tetrahydrouridine (THU), a potent CD inhibitor, has been applied preclinically and clinically as a modulator of cytidine analogue metabolism. However, THU is only 20% orally bioavailable, which limits its preclinical evaluation and clinical use. Therefore, we characterized THU pharmacokinetics after the administration to mice of the more lipophilic pro-drug triacetyl-THU (taTHU).
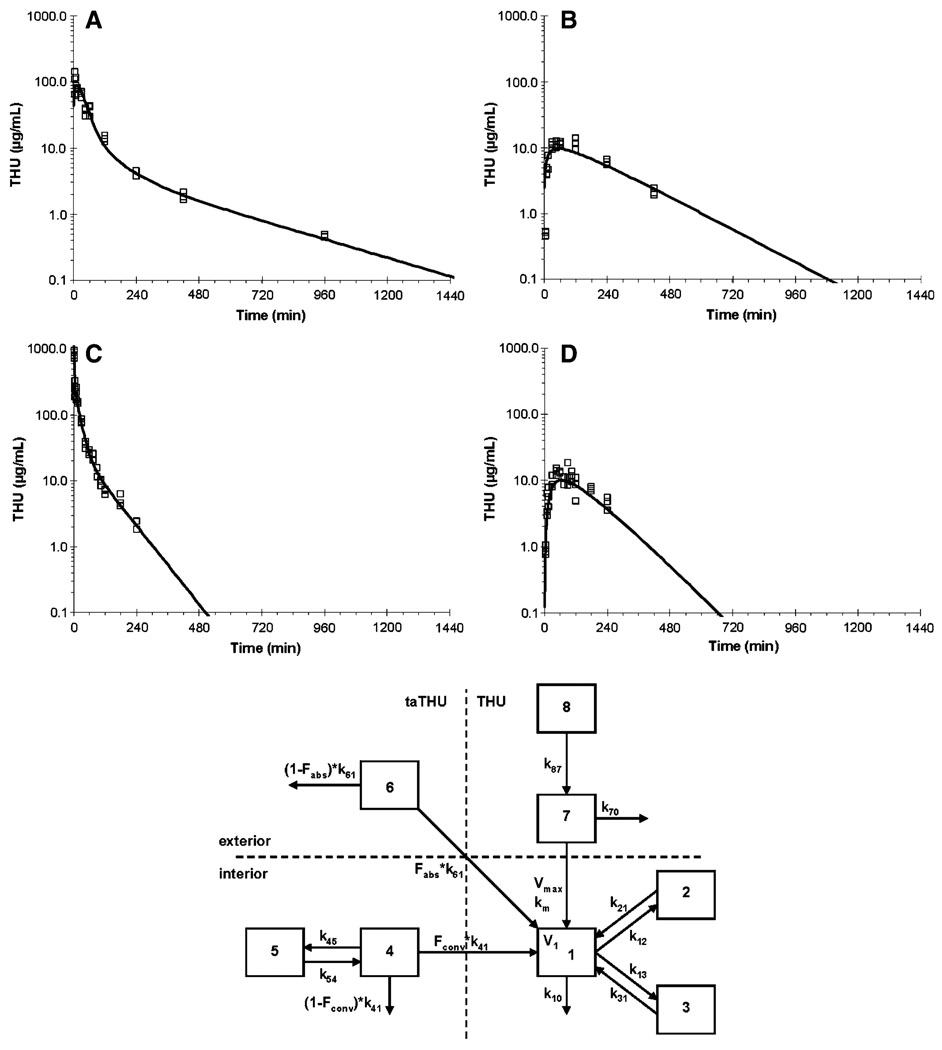
Methods: Mice were dosed with 150 mg/kg taTHU i.v. or p.o. Plasma and urine THU concentrations were quantitated with a validated LC-MS/MS assay. Plasma and urine pharmacokinetic parameters were calculated non-compartmentally and compartmentally.
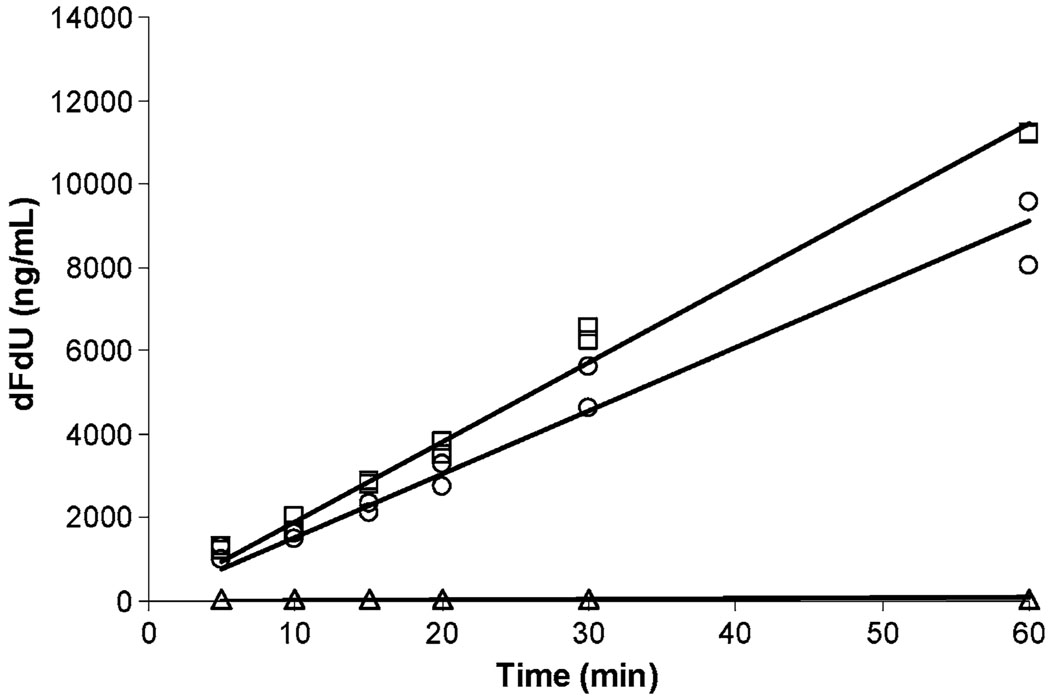
Results: taTHU did not inhibit CD. THU, after 150 mg/kg taTHU i.v., had a 235-min terminal half-life and produced plasma THU concentrations >1 μg/mL, the concentration shown to inhibit CD, for 10 h. Renal excretion accounted for 40-55% of the i.v. taTHU dose, 6-12% of the p.o. taTHU dose. A two-compartment model of taTHU generating THU fitted the i.v. taTHU data best. taTHU, at 150 mg/kg p.o., produced a concentration versus time profile with a plateau of approximately 10 μg/mL from 0.5-2 h, followed by a decline with a 122-min half-life. Approximately 68% of i.v. taTHU is converted to THU. Approximately 30% of p.o. taTHU reaches the systemic circulation as THU.
Conclusions: The availability of THU after p.o. taTHU is 30%, when compared to the 20% achieved with p.o. THU. These data will support the clinical studies of taTHU.
Conflict of interest statement
Figures
Similar articles
-
Plasma pharmacokinetics and oral bioavailability of 3,4,5,6-tetrahydrouridine, a cytidine deaminase inhibitor, in mice.Cancer Chemother Pharmacol. 2008 Aug;62(3):457-64. doi: 10.1007/s00280-007-0625-2. Epub 2007 Nov 15. Cancer Chemother Pharmacol. 2008. PMID: 18008070 Free PMC article.
-
Modulation of gemcitabine (2',2'-difluoro-2'-deoxycytidine) pharmacokinetics, metabolism, and bioavailability in mice by 3,4,5,6-tetrahydrouridine.Clin Cancer Res. 2008 Jun 1;14(11):3529-35. doi: 10.1158/1078-0432.CCR-07-4885. Clin Cancer Res. 2008. PMID: 18519786
-
Oral and intravenous pharmacokinetics of 5-fluoro-2'-deoxycytidine and THU in cynomolgus monkeys and humans.Cancer Chemother Pharmacol. 2015 Oct;76(4):803-11. doi: 10.1007/s00280-015-2857-x. Epub 2015 Sep 1. Cancer Chemother Pharmacol. 2015. PMID: 26321472 Free PMC article. Clinical Trial.
-
Gemcitabine: metabolism, mechanisms of action, and self-potentiation.Semin Oncol. 1995 Aug;22(4 Suppl 11):3-10. Semin Oncol. 1995. PMID: 7481842 Review.
-
[Clinical pharmacology of nucleoside analogues].Bull Cancer. 2002 Aug;89 Spec No:S71-5. Bull Cancer. 2002. PMID: 12449033 Review. French.
Cited by
-
Formation of active products of benzaldehyde dimethane sulfonate (NSC 281612, DMS612) in human blood and plasma and their activity against renal cell carcinoma lines.Cancer Chemother Pharmacol. 2013 Jan;71(1):73-83. doi: 10.1007/s00280-012-1980-1. Epub 2012 Oct 5. Cancer Chemother Pharmacol. 2013. PMID: 23053264 Free PMC article.
-
The Emerging Role of Cytidine Deaminase in Human Diseases: A New Opportunity for Therapy?Mol Ther. 2020 Feb 5;28(2):357-366. doi: 10.1016/j.ymthe.2019.11.026. Epub 2019 Dec 6. Mol Ther. 2020. PMID: 31870623 Free PMC article. Review.
-
Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE).PLoS Genet. 2011 Mar;7(3):e1002035. doi: 10.1371/journal.pgen.1002035. Epub 2011 Mar 31. PLoS Genet. 2011. PMID: 21483760 Free PMC article.
-
Targeting Chemoresistance in Advanced Bladder Cancers with a Novel Adjuvant Strategy.Mol Cancer Ther. 2024 May 30:10.1158/1535-7163.MCT-23-0806. doi: 10.1158/1535-7163.MCT-23-0806. Online ahead of print. Mol Cancer Ther. 2024. PMID: 38814440
-
Attempts to remodel the pathways of gemcitabine metabolism: Recent approaches to overcoming tumours with acquired chemoresistance.Cancer Drug Resist. 2020 Oct 12;3(4):819-831. doi: 10.20517/cdr.2020.39. eCollection 2020. Cancer Drug Resist. 2020. PMID: 35582220 Free PMC article. Review.
References
-
- Akaike H. A Bayesian extension of the minimal AIC procedures of autoregressive model fitting. Biometrika. 1979;66:237–242.
-
- Ashour OM, Naguib FN, el Kouni MH. 5-(m-Benzyloxy-benzyl) barbituric acid acyclonucleoside, a uridine phosphorylase inhibitor, and 2′, 3′, 5′-tri-O-acetyluridine, a prodrug of uridine, as modulators of plasma uridine concentration. Implications for chemotherapy. Biochem Pharmacol. 1996;51:1601–1611. - PubMed
-
- Beumer JH, Eiseman JL, Parise RA, Joseph E, Covey JM, Egorin MJ. Modulation of gemcitabine (2′, 2′-difluoro-2′-deoxycytidine) pharmacokinetics, metabolism, and bioavailability in mice by 3, 4, 5, 6-tetrahydrouridine. Clin Cancer Res. 2008;14:3529–3535. - PubMed
-
- Beumer JH, Eiseman JL, Parise RA, Joseph E, Holleran JL, Covey JM, Egorin MJ. Pharmacokinetics, metabolism, and oral bioavailability of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine in mice. Clin Cancer Res. 2006;12:7483–7491. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
