

A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling
- PMID: 20444259
- PMCID: PMC2877694
- DOI: 10.1186/1471-2164-11-282
A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling
Abstract
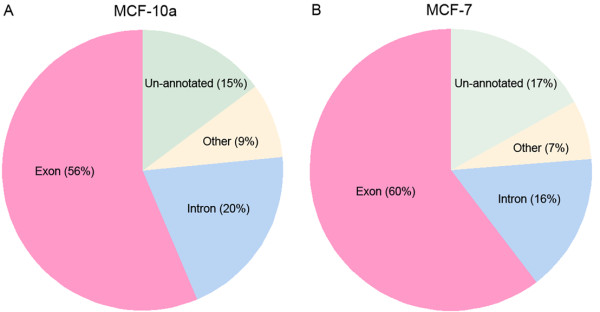
Background: RNA-Seq exploits the rapid generation of gigabases of sequence data by Massively Parallel Nucleotide Sequencing, allowing for the mapping and digital quantification of whole transcriptomes. Whilst previous comparisons between RNA-Seq and microarrays have been performed at the level of gene expression, in this study we adopt a more fine-grained approach. Using RNA samples from a normal human breast epithelial cell line (MCF-10a) and a breast cancer cell line (MCF-7), we present a comprehensive comparison between RNA-Seq data generated on the Applied Biosystems SOLiD platform and data from Affymetrix Exon 1.0ST arrays. The use of Exon arrays makes it possible to assess the performance of RNA-Seq in two key areas: detection of expression at the granularity of individual exons, and discovery of transcription outside annotated loci.
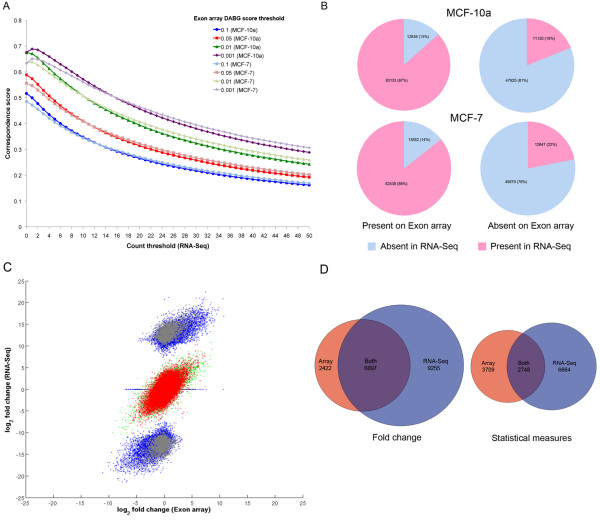
Results: We found a high degree of correspondence between the two platforms in terms of exon-level fold changes and detection. For example, over 80% of exons detected as expressed in RNA-Seq were also detected on the Exon array, and 91% of exons flagged as changing from Absent to Present on at least one platform had fold-changes in the same direction. The greatest detection correspondence was seen when the read count threshold at which to flag exons Absent in the SOLiD data was set to t<1 suggesting that the background error rate is extremely low in RNA-Seq. We also found RNA-Seq more sensitive to detecting differentially expressed exons than the Exon array, reflecting the wider dynamic range achievable on the SOLiD platform. In addition, we find significant evidence of novel protein coding regions outside known exons, 93% of which map to Exon array probesets, and are able to infer the presence of thousands of novel transcripts through the detection of previously unreported exon-exon junctions.
Conclusions: By focusing on exon-level expression, we present the most fine-grained comparison between RNA-Seq and microarrays to date. Overall, our study demonstrates that data from a SOLiD RNA-Seq experiment are sufficient to generate results comparable to those produced from Affymetrix Exon arrays, even using only a single replicate from each platform, and when presented with a large genome.
Figures
Similar articles
-
A systematic comparison and evaluation of high density exon arrays and RNA-seq technology used to unravel the peripheral blood transcriptome of sickle cell disease.BMC Med Genomics. 2012 Jun 29;5:28. doi: 10.1186/1755-8794-5-28. BMC Med Genomics. 2012. PMID: 22747986 Free PMC article.
-
Gene expression and isoform variation analysis using Affymetrix Exon Arrays.BMC Genomics. 2008 Nov 7;9:529. doi: 10.1186/1471-2164-9-529. BMC Genomics. 2008. PMID: 18990248 Free PMC article.
-
A comparison of RNA-seq and exon arrays for whole genome transcription profiling of the L5 spinal nerve transection model of neuropathic pain in the rat.Mol Pain. 2014 Jan 28;10:7. doi: 10.1186/1744-8069-10-7. Mol Pain. 2014. PMID: 24472155 Free PMC article.
-
Transcribed dark matter: meaning or myth?Hum Mol Genet. 2010 Oct 15;19(R2):R162-8. doi: 10.1093/hmg/ddq362. Epub 2010 Aug 25. Hum Mol Genet. 2010. PMID: 20798109 Free PMC article. Review.
-
Introduction to sequencing the brain transcriptome.Int Rev Neurobiol. 2014;116:1-19. doi: 10.1016/B978-0-12-801105-8.00001-1. Int Rev Neurobiol. 2014. PMID: 25172469 Free PMC article. Review.
Cited by
-
Accurate diagnostics for Bovine tuberculosis based on high-throughput sequencing.PLoS One. 2012;7(11):e50147. doi: 10.1371/journal.pone.0050147. Epub 2012 Nov 30. PLoS One. 2012. PMID: 23226242 Free PMC article.
-
Next-generation sequencing facilitates quantitative analysis of wild-type and Nrl(-/-) retinal transcriptomes.Mol Vis. 2011;17:3034-54. Epub 2011 Nov 23. Mol Vis. 2011. PMID: 22162623 Free PMC article.
-
Omics Application of Bio-Hydrogen Production Through Green Alga Chlamydomonas reinhardtii.Front Bioeng Biotechnol. 2019 Aug 21;7:201. doi: 10.3389/fbioe.2019.00201. eCollection 2019. Front Bioeng Biotechnol. 2019. PMID: 31497598 Free PMC article. Review.
-
Nutrigenomics in honey bees: digital gene expression analysis of pollen's nutritive effects on healthy and varroa-parasitized bees.BMC Genomics. 2011 Oct 10;12:496. doi: 10.1186/1471-2164-12-496. BMC Genomics. 2011. PMID: 21985689 Free PMC article.
-
From RNA-seq reads to differential expression results.Genome Biol. 2010;11(12):220. doi: 10.1186/gb-2010-11-12-220. Epub 2010 Dec 22. Genome Biol. 2010. PMID: 21176179 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
