

Novel protein kinase D inhibitors cause potent arrest in prostate cancer cell growth and motility
- PMID: 20444281
- PMCID: PMC2873968
- DOI: 10.1186/1472-6769-10-5
Novel protein kinase D inhibitors cause potent arrest in prostate cancer cell growth and motility
Abstract
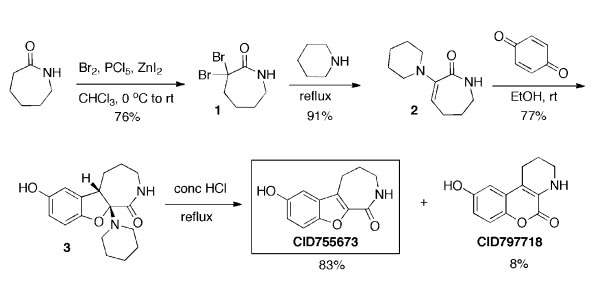
Background: Protein kinase D (PKD) has been implicated in a wide range of cellular processes and pathological conditions including cancer. However, targeting PKD therapeutically and dissecting PKD-mediated cellular responses remains difficult due to lack of a potent and selective inhibitor. Previously, we identified a novel pan-PKD inhibitor, CID755673, with potency in the upper nanomolar range and high selectivity for PKD. In an effort to further enhance its selectivity and potency for potential in vivo application, small molecule analogs of CID755673 were generated by modifying both the core structure and side-chains.
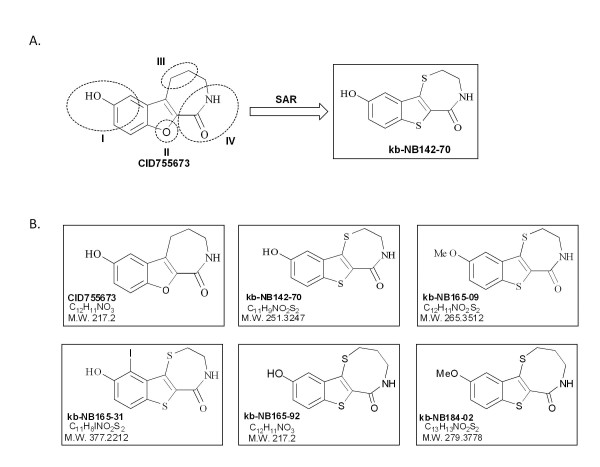
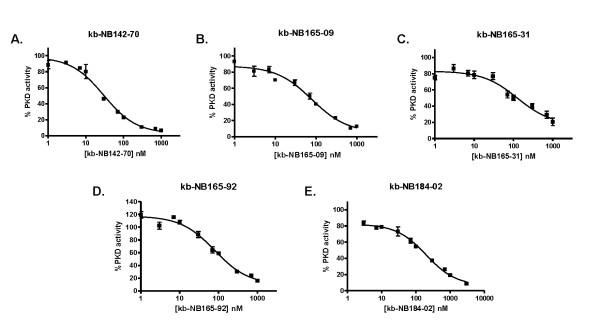
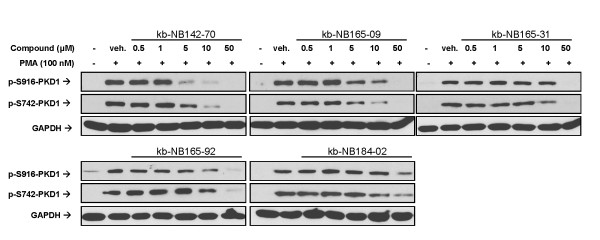
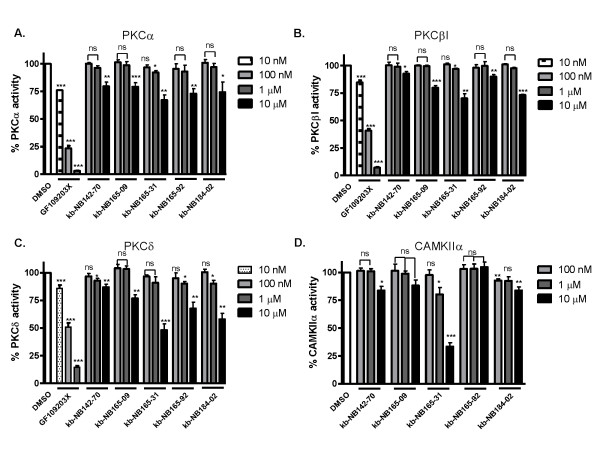
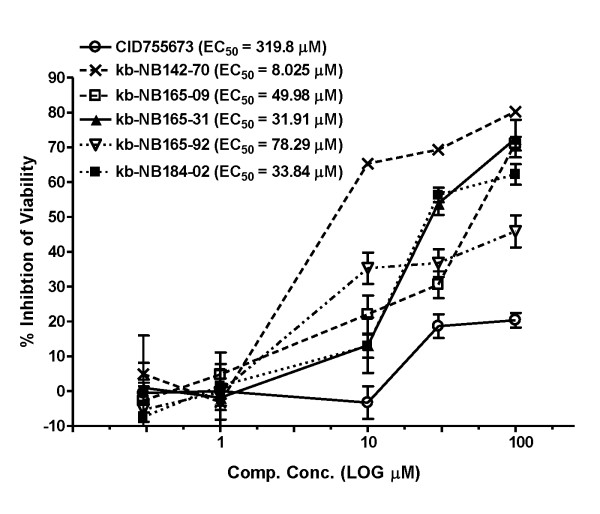
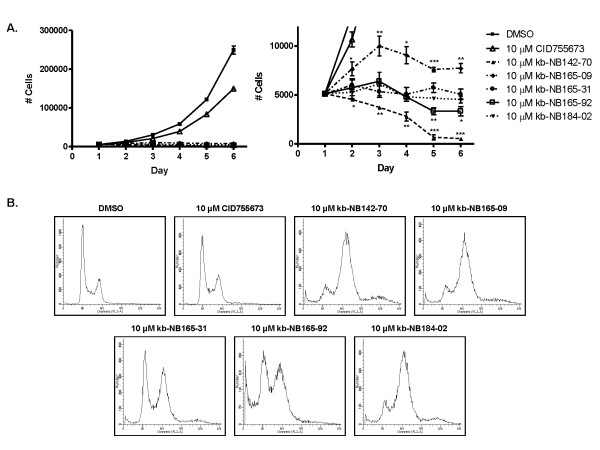
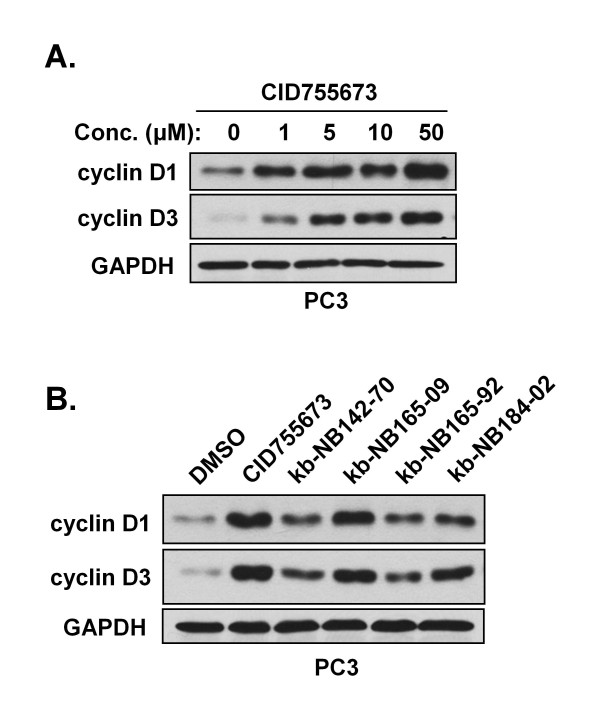
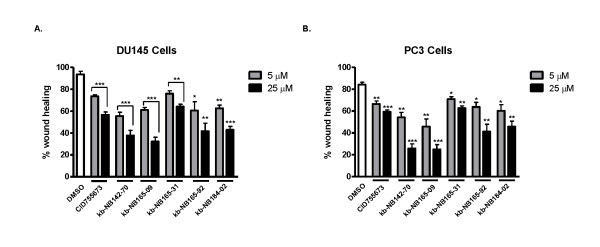
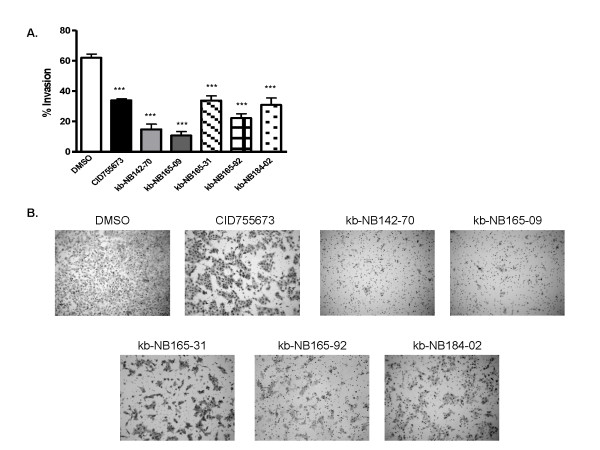
Results: After initial activity screening, five analogs with equal or greater potencies as CID755673 were chosen for further analysis: kb-NB142-70, kb-NB165-09, kb-NB165-31, kb-NB165-92, and kb-NB184-02. Our data showed that modifications to the aromatic core structure in particular significantly increased potency while retaining high specificity for PKD. When tested in prostate cancer cells, all compounds inhibited PMA-induced autophosphorylation of PKD1, with kb-NB142-70 being most active. Importantly, these analogs caused a dramatic arrest in cell proliferation accompanying elevated cytotoxicity when applied to prostate cancer cells. Cell migration and invasion were also inhibited by these analogs with varying potencies that correlated to their cellular activity.
Conclusions: Throughout the battery of experiments, the compounds kb-NB142-70 and kb-NB165-09 emerged as the most potent and specific analogs in vitro and in cells. These compounds are undergoing further testing for their effectiveness as pharmacological tools for dissecting PKD function and as potential anti-cancer agents in the treatment of prostate cancer.
Figures
Similar articles
-
Synthesis and Structure-Activity Relationships of Benzothienothiazepinone Inhibitors of Protein Kinase D.ACS Med Chem Lett. 2011 Feb 14;2(2):154-159. doi: 10.1021/ml100230n. ACS Med Chem Lett. 2011. PMID: 21617763 Free PMC article.
-
In vitro cytotoxicity, pharmacokinetics, tissue distribution, and metabolism of small-molecule protein kinase D inhibitors, kb-NB142-70 and kb-NB165-09, in mice bearing human cancer xenografts.Cancer Chemother Pharmacol. 2013 Feb;71(2):331-44. doi: 10.1007/s00280-012-2010-z. Epub 2012 Oct 30. Cancer Chemother Pharmacol. 2013. PMID: 23108699 Free PMC article.
-
Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone.J Biol Chem. 2008 Nov 28;283(48):33516-26. doi: 10.1074/jbc.M805358200. Epub 2008 Sep 30. J Biol Chem. 2008. PMID: 18829454 Free PMC article.
-
Small-Molecule Inhibitor Targeting Protein Kinase D: A Potential Therapeutic Strategy.Front Oncol. 2021 Jun 24;11:680221. doi: 10.3389/fonc.2021.680221. eCollection 2021. Front Oncol. 2021. PMID: 34249722 Free PMC article. Review.
-
PKD at the crossroads of DAG and PKC signaling.Trends Pharmacol Sci. 2006 Jun;27(6):317-23. doi: 10.1016/j.tips.2006.04.003. Epub 2006 May 6. Trends Pharmacol Sci. 2006. PMID: 16678913 Review.
Cited by
-
Protein Kinase D2 Modulates Cell Cycle By Stabilizing Aurora A Kinase at Centrosomes.Mol Cancer Res. 2018 Nov;16(11):1785-1797. doi: 10.1158/1541-7786.MCR-18-0641. Epub 2018 Jul 17. Mol Cancer Res. 2018. PMID: 30018032 Free PMC article.
-
Synthesis and Structure-Activity Relationships of Benzothienothiazepinone Inhibitors of Protein Kinase D.ACS Med Chem Lett. 2011 Feb 14;2(2):154-159. doi: 10.1021/ml100230n. ACS Med Chem Lett. 2011. PMID: 21617763 Free PMC article.
-
An Evolutionarily Conserved PLC-PKD-TFEB Pathway for Host Defense.Cell Rep. 2016 May 24;15(8):1728-42. doi: 10.1016/j.celrep.2016.04.052. Epub 2016 May 12. Cell Rep. 2016. PMID: 27184844 Free PMC article.
-
Endothelial Protein kinase D1 is a major regulator of post-traumatic hyperinflammation.Front Immunol. 2023 Mar 2;14:1093022. doi: 10.3389/fimmu.2023.1093022. eCollection 2023. Front Immunol. 2023. PMID: 36936923 Free PMC article.
-
Hope and fear for interferon: the receptor-centric outlook on the future of interferon therapy.J Interferon Cytokine Res. 2013 Apr;33(4):211-25. doi: 10.1089/jir.2012.0117. J Interferon Cytokine Res. 2013. PMID: 23570388 Free PMC article. Review.
References
-
- Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K. PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem. 1994;269(8):6140–6148. - PubMed
-
- Sturany S, Van Lint J, Muller F, Wilda M, Hameister H, Hocker M, Brey A, Gern U, Vandenheede J, Gress T. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J Biol Chem. 2001;276(5):3310–3318. doi: 10.1074/jbc.M008719200. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Chemical Information
Molecular Biology Databases
