

e-LEA3D: a computational-aided drug design web server
- PMID: 20444867
- PMCID: PMC2896156
- DOI: 10.1093/nar/gkq322
e-LEA3D: a computational-aided drug design web server
Abstract
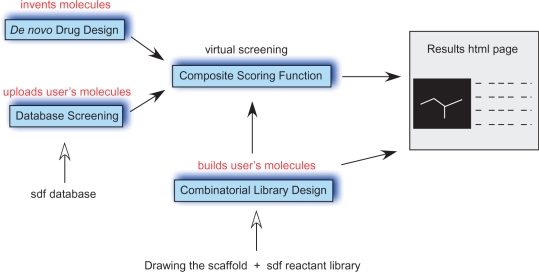
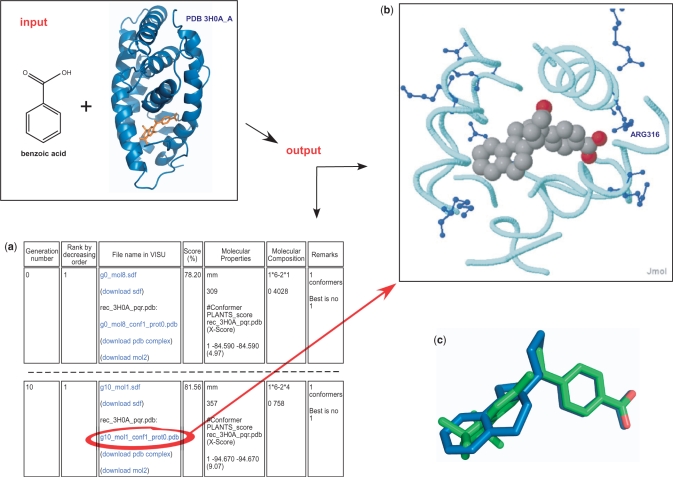
e-LEA3D web server integrates three complementary tools to perform computer-aided drug design based on molecular fragments. In drug discovery projects, there is a considerable interest in identifying novel and diverse molecular scaffolds to enhance chances of success. The de novo drug design tool is used to invent new ligands to optimize a user-specified scoring function. The composite scoring function includes both structure- and ligand-based evaluations. The de novo approach is an alternative to a blind virtual screening of large compound collections. A heuristic based on a genetic algorithm rapidly finds which fragments or combination of fragments fit a QSAR model or the binding site of a protein. While the approach is ideally suited for scaffold-hopping, this module also allows a scan for possible substituents to a user-specified scaffold. The second tool offers a traditional virtual screening and filtering of an uploaded library of compounds. The third module addresses the combinatorial library design that is based on a user-drawn scaffold and reactants coming, for example, from a chemical supplier. The e-LEA3D server is available at: http://bioinfo.ipmc.cnrs.fr/lea.html.
Figures
References
-
- Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–861. - PubMed
-
- Triggle DJ. The chemist as astronaut: searching for biologically useful space in the chemical universe. Biochem. Pharmacol. 2009;78:217–223. - PubMed
-
- Blum LC, Reymond JL. 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc. 2009;131:8732–8733. - PubMed
-
- Hartenfeller M, Proschak E, Schuller A, Schneider G. Concept of combinatorial de novo design of drug-like molecules by particle swarm optimization. Chem. Biol. Drug Des. 2008;72:16–26. - PubMed
-
- Masek BB, Shen L, Smith KM, Pearlman RS. Sharing chemical information without sharing chemical structure. J. Chem. Inf. Model. 2008;48:256–261. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
