

Processing of lagging-strand intermediates in vitro by herpes simplex virus type 1 DNA polymerase
- PMID: 20444887
- PMCID: PMC2897638
- DOI: 10.1128/JVI.01875-09
Processing of lagging-strand intermediates in vitro by herpes simplex virus type 1 DNA polymerase
Abstract
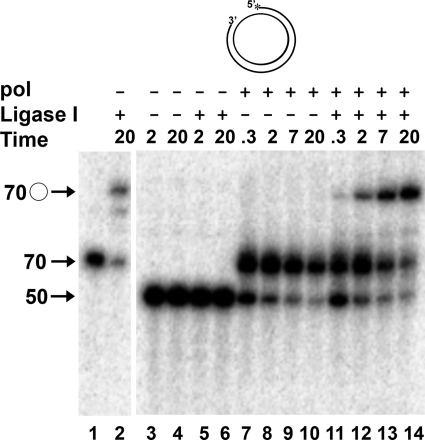
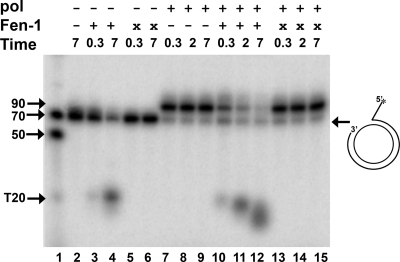
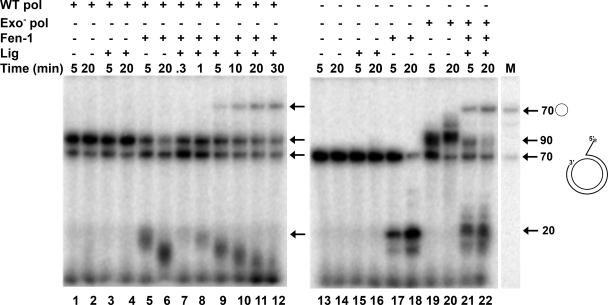
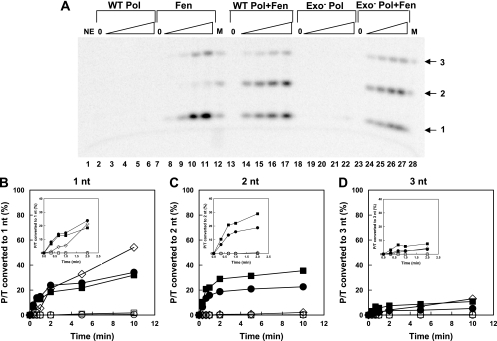
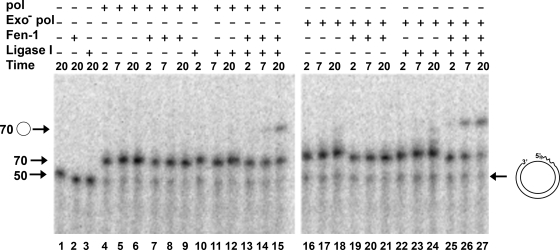
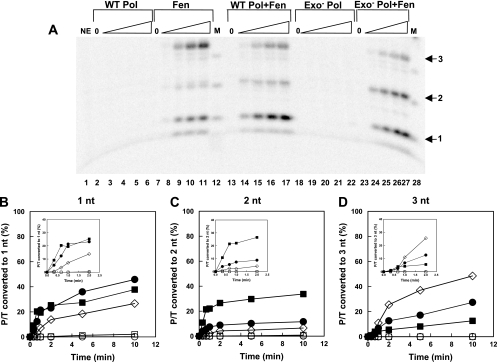
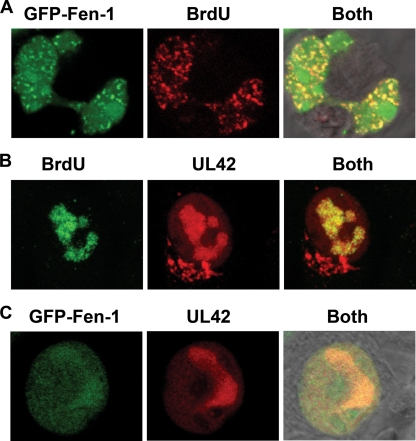
The processing of lagging-strand intermediates has not been demonstrated in vitro for herpes simplex virus type 1 (HSV-1). Human flap endonuclease-1 (Fen-1) was examined for its ability to produce ligatable products with model lagging-strand intermediates in the presence of the wild-type or exonuclease-deficient (exo(-)) HSV-1 DNA polymerase (pol). Primer/templates were composed of a minicircle single-stranded DNA template annealed to primers that contained 5' DNA flaps or 5' annealed DNA or RNA sequences. Gapped DNA primer/templates were extended but not significantly strand displaced by the wild-type HSV-1 pol, although significant strand displacement was observed with exo(-) HSV-1 pol. Nevertheless, the incubation of primer/templates containing 5' flaps with either wild-type or exo(-) HSV-1 pol and Fen-1 led to the efficient production of nicks that could be sealed with DNA ligase I. Both polymerases stimulated the nick translation activity of Fen-1 on DNA- or RNA-containing primer/templates, indicating that the activities were coordinated. Further evidence for Fen-1 involvement in HSV-1 DNA synthesis is suggested by the ability of a transiently expressed green fluorescent protein fusion with Fen-1 to accumulate in viral DNA replication compartments in infected cells and by the ability of endogenous Fen-1 to coimmunoprecipitate with an essential viral DNA replication protein in HSV-1-infected cells.
Figures
References
-
- Ayyagari, R., X. V. Gomes, D. A. Gordenin, and P. M. J. Burgers. 2003. Okazaki fragment maturation in yeast. I. Distribution of functions between Fen1 and Dna2. J. Biol. Chem. 278:1618-1625. - PubMed
-
- Bae, S.-H., and Y.-S. Seo. 2000. Characterization of the enzymatic properties of the yeast Dna2 helicase/endonuclease suggests a new model for Okazaki fragment processing. J. Biol. Chem. 275:38022-38031. - PubMed
-
- Bentley, D. J., C. Harrison, A.-M. Ketchen, N. J. Redhead, K. Samuel, M. Waterfall, J. D. Ansell, and D. W. Melton. 2002. DNA ligase I null mouse cells show normal DNA repair activity but altered DNA replication and reduced genome stability. J. Cell Sci. 115:1551-1561. - PubMed
-
- Biswal, N., B. K. Murry, and M. Benyish-Melnick. 1974. Ribonucleotides in newly synthesized DNA of herpes simplex virus. Virology 69:87-99. - PubMed
-
- Boehmer, P. E., and A. V. Nimonkar. 2003. Herpes virus replication. IUBMB Life 55:13-22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
