

Endothelin signalling regulates volume-sensitive Cl- current via NADPH oxidase and mitochondrial reactive oxygen species
- PMID: 20444986
- PMCID: PMC2936123
- DOI: 10.1093/cvr/cvq125
Endothelin signalling regulates volume-sensitive Cl- current via NADPH oxidase and mitochondrial reactive oxygen species
Abstract
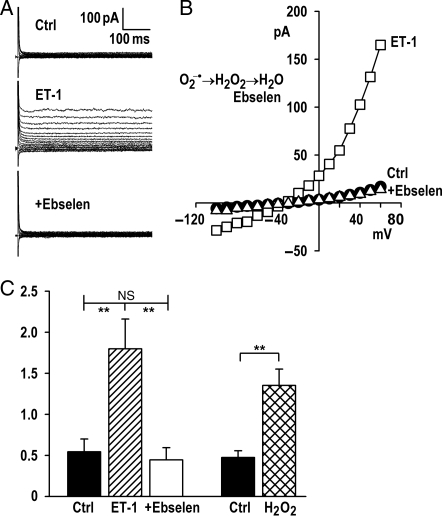
Aims: We assessed regulation of volume-sensitive Cl(-) current (I(Cl,swell)) by endothelin-1 (ET-1) and characterized the signalling pathway responsible for its activation in rabbit atrial and ventricular myocytes.
Methods and results: ET-1 elicited I(Cl,swell) under isosmotic conditions. Outwardly rectified Cl(-) current was blocked by the I(Cl,swell)-selective inhibitor DCPIB or osmotic shrinkage and involved ET(A) but not ET(B) receptors. ET-1-induced current was abolished by inhibiting epidermal growth factor receptor (EGFR) kinase or phosphoinositide-3-kinase (PI-3K), indicating that these kinases were downstream. Regarding upstream events, activation of I(Cl,swell) by osmotic swelling or angiotensin II (AngII) was suppressed by ET(A) blockade, whereas AngII AT(1) receptor blockade failed to alter ET-1-induced current. Reactive oxygen species (ROS) produced by NADPH oxidase (NOX) stimulate I(Cl,swell). As expected, blockade of NOX suppressed ET-1-induced I(Cl,swell), but blockade of mitochondrial ROS production with rotenone also suppressed I(Cl,swell). I(Cl,swell) was activated by augmenting complex III ROS production with antimycin A or diazoxide; in this case, I(Cl,swell) was insensitive to NOX inhibitors, indicating that mitochondria were downstream from NOX. ROS generation in HL-1 cardiomyocytes measured by flow cytometry confirmed the electrophysiological findings. ET-1-induced ROS production was inhibited by blocking either NOX or mitochondrial complex I, whereas complex III-induced ROS production was insensitive to NOX blockade.
Conclusion: ET-1-ET(A) signalling activated I(Cl,swell) via EGFR kinase, PI-3K, and NOX ROS production, which triggered mitochondrial ROS production. ET(A) receptors were downstream effectors when I(Cl,swell) was elicited by osmotic swelling or AngII. These data suggest that ET-1-induced ROS-dependent I(Cl,swell) is likely to participate in multiple physiological and pathophysiological processes.
Figures






Similar articles
-
Regulation of swelling-activated Cl(-) current by angiotensin II signalling and NADPH oxidase in rabbit ventricle.Cardiovasc Res. 2008 Jan;77(1):73-80. doi: 10.1093/cvr/cvm031. Epub 2007 Oct 4. Cardiovasc Res. 2008. PMID: 18006461 Free PMC article.
-
HIV protease inhibitors elicit volume-sensitive Cl- current in cardiac myocytes via mitochondrial ROS.J Mol Cell Cardiol. 2010 Nov;49(5):746-52. doi: 10.1016/j.yjmcc.2010.08.013. Epub 2010 Aug 22. J Mol Cell Cardiol. 2010. PMID: 20736017 Free PMC article.
-
EGFR kinase regulates volume-sensitive chloride current elicited by integrin stretch via PI-3K and NADPH oxidase in ventricular myocytes.J Gen Physiol. 2006 Mar;127(3):237-51. doi: 10.1085/jgp.200509366. Epub 2006 Feb 13. J Gen Physiol. 2006. PMID: 16505146 Free PMC article.
-
Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species.Biochim Biophys Acta. 2010 Jun-Jul;1797(6-7):897-906. doi: 10.1016/j.bbabio.2010.01.032. Epub 2010 Feb 1. Biochim Biophys Acta. 2010. PMID: 20122895 Review.
-
Swelling-activated chloride channels in cardiac physiology and pathophysiology.Prog Biophys Mol Biol. 2003 May-Jul;82(1-3):25-42. doi: 10.1016/s0079-6107(03)00003-8. Prog Biophys Mol Biol. 2003. PMID: 12732266 Review.
Cited by
-
Phenomics of cardiac chloride channels.Compr Physiol. 2013 Apr;3(2):667-92. doi: 10.1002/cphy.c110014. Compr Physiol. 2013. PMID: 23720326 Free PMC article. Review.
-
Cross talk between mitochondria and NADPH oxidases.Free Radic Biol Med. 2011 Oct 1;51(7):1289-301. doi: 10.1016/j.freeradbiomed.2011.06.033. Epub 2011 Jul 6. Free Radic Biol Med. 2011. PMID: 21777669 Free PMC article. Review.
-
LRRC8/VRAC channels exhibit a noncanonical permeability to glutathione, which modulates epithelial-to-mesenchymal transition (EMT).Cell Death Dis. 2019 Dec 5;10(12):925. doi: 10.1038/s41419-019-2167-z. Cell Death Dis. 2019. PMID: 31804464 Free PMC article.
-
Noradrenaline up-regulates volume-regulated chloride current by PKA-independent cAMP/exchange protein activated by cAMP pathway in human atrial myocytes.Br J Pharmacol. 2018 Aug;175(16):3422-3432. doi: 10.1111/bph.14392. Epub 2018 Jul 8. Br J Pharmacol. 2018. PMID: 29900525 Free PMC article.
-
Stress-induced modulation of volume-regulated anions channels in human alveolar carcinoma cells.Physiol Rep. 2018 Sep;6(19):e13869. doi: 10.14814/phy2.13869. Physiol Rep. 2018. PMID: 30318853 Free PMC article.
References
-
- Brunner F, Bras-Silva C, Cerdeira AS, Leite-Moreira AF. Cardiovascular endothelins: essential regulators of cardiovascular homeostasis. Pharmacol Ther. 2006;111:508–531. - PubMed
-
- Ono K, Tsujimoto G, Sakamoto A, Eto K, Masaki T, Ozaki Y, et al. Endothelin-A receptor mediates cardiac inhibition by regulating calcium and potassium currents. Nature. 1994;370:301–304. doi:10.1038/370301a0. - DOI - PubMed
-
- Kiesecker C, Zitron E, Scherer D, Lueck S, Bloehs R, Scholz EP, et al. Regulation of cardiac inwardly rectifying potassium current IK1 and Kir2.x channels by endothelin-1. J Mol Med. 2006;84:46–56. doi:10.1007/s00109-005-0707-8. - DOI - PubMed
-
- Du XY, Sorota S. Cardiac swelling-induced chloride current is enhanced by endothelin. J Cardiovasc Pharmacol. 2000;35:769–776. doi:10.1097/00005344-200005000-00014. - DOI - PubMed
-
- Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl− current elicited by β1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol. 2004;124:273–287. doi:10.1085/jgp.200409040. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
