

The distribution of inverted repeat sequences in the Saccharomyces cerevisiae genome
- PMID: 20446088
- PMCID: PMC2908449
- DOI: 10.1007/s00294-010-0302-6
The distribution of inverted repeat sequences in the Saccharomyces cerevisiae genome
Abstract
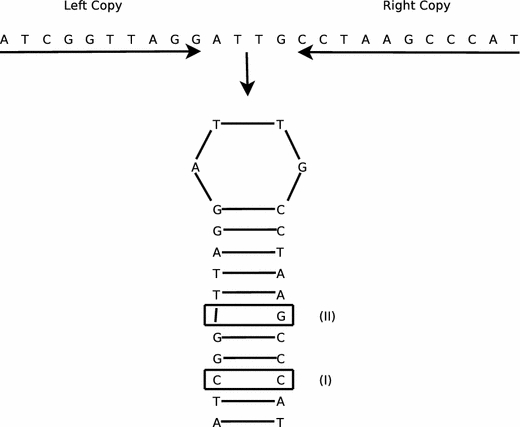
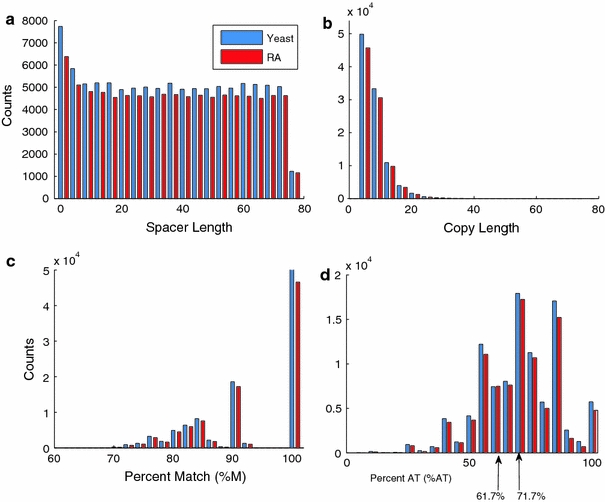
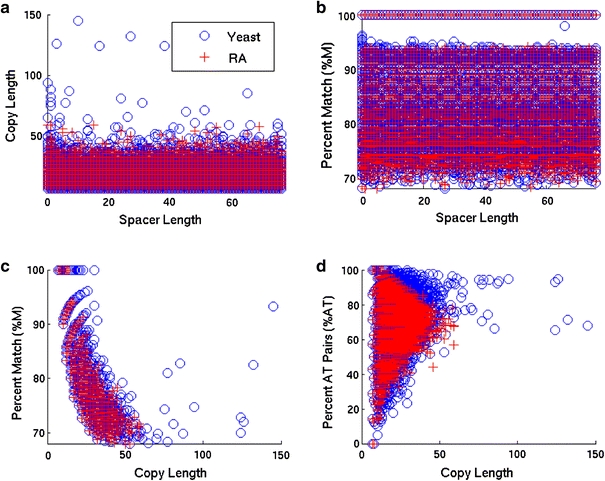
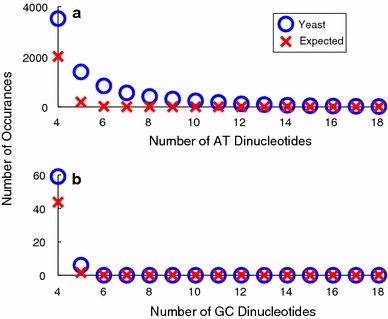
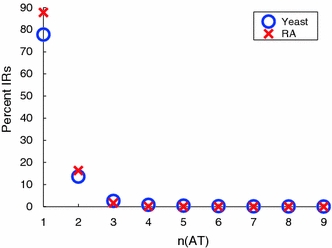
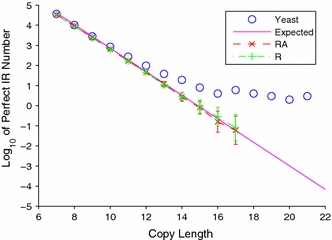
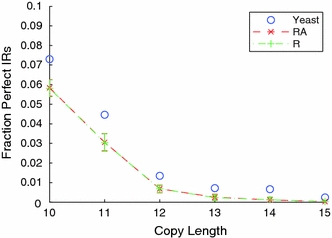
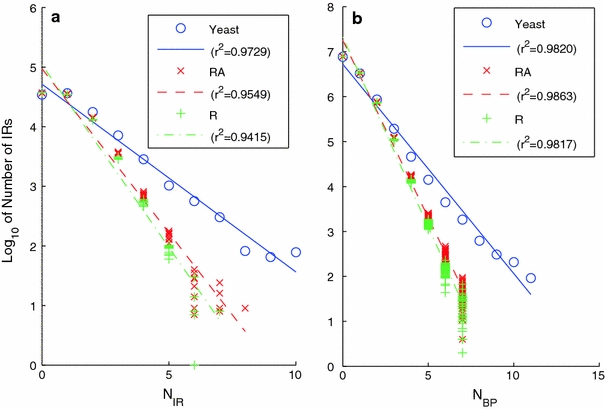
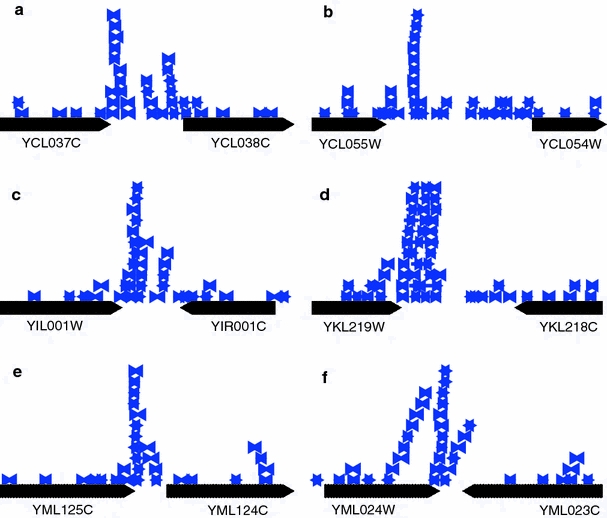
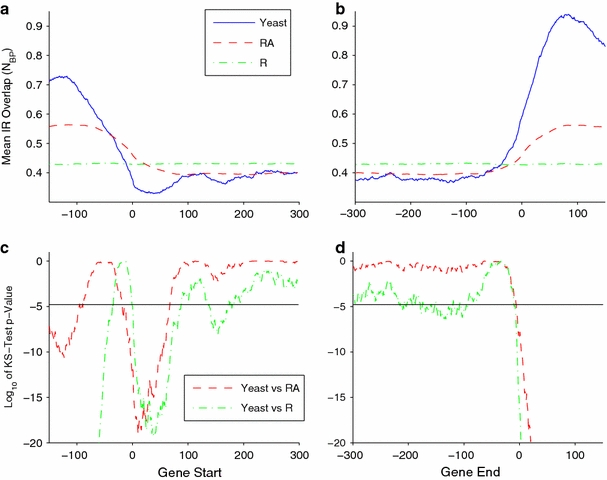
Although a variety of possible functions have been proposed for inverted repeat sequences (IRs), it is not known which of them might occur in vivo. We investigate this question by assessing the distributions and properties of IRs in the Saccharomyces cerevisiae (SC) genome. Using the IRFinder algorithm we detect 100,514 IRs having copy length greater than 6 bp and spacer length less than 77 bp. To assess statistical significance we also determine the IR distributions in two types of randomization of the S. cerevisiae genome. We find that the S. cerevisiae genome is significantly enriched in IRs relative to random. The S. cerevisiae IRs are significantly longer and contain fewer imperfections than those from the randomized genomes, suggesting that processes to lengthen and/or correct errors in IRs may be operative in vivo. The S. cerevisiae IRs are highly clustered in intergenic regions, while their occurrence in coding sequences is consistent with random. Clustering is stronger in the 3' flanks of genes than in their 5' flanks. However, the S. cerevisiae genome is not enriched in those IRs that would extrude cruciforms, suggesting that this is not a common event. Various explanations for these results are considered.
Figures
References
-
- Achez G, Coissac E, Viari A, Netter P. Analysis of intrachromosomal duplications in yeast Saccharomyces cerevisiae: a possible model for their origin. Mol Biol Evol. 2000;17:1268–1275. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
