

Differences in presentation and progression between severe FIC1 and BSEP deficiencies
- PMID: 20447715
- PMCID: PMC3042805
- DOI: 10.1016/j.jhep.2010.01.034
Differences in presentation and progression between severe FIC1 and BSEP deficiencies
Abstract
Background & aims: Progressive familial intrahepatic cholestasis (PFIC) with normal serum levels of gamma-glutamyltranspeptidase can result from mutations in ATP8B1 (encoding familial intrahepatic cholestasis 1 [FIC1]) or ABCB11 (encoding bile salt export pump [BSEP]). We evaluated clinical and laboratory features of disease in patients diagnosed with PFIC, who carried mutations in ATP8B1 (FIC1 deficiency) or ABCB11 (BSEP deficiency). Our goal was to identify features that distinguish presentation and course of these two disorders, thus facilitating diagnosis and elucidating the differing consequences of ATP8B1 and ABCB11 mutations.
Methods: A retrospective multi-center study was conducted, using questionnaires and chart review. Available clinical and biochemical data from 145 PFIC patients with mutations in either ATP8B1 (61 "FIC1 patients") or ABCB11 (84 "BSEP patients") were evaluated.
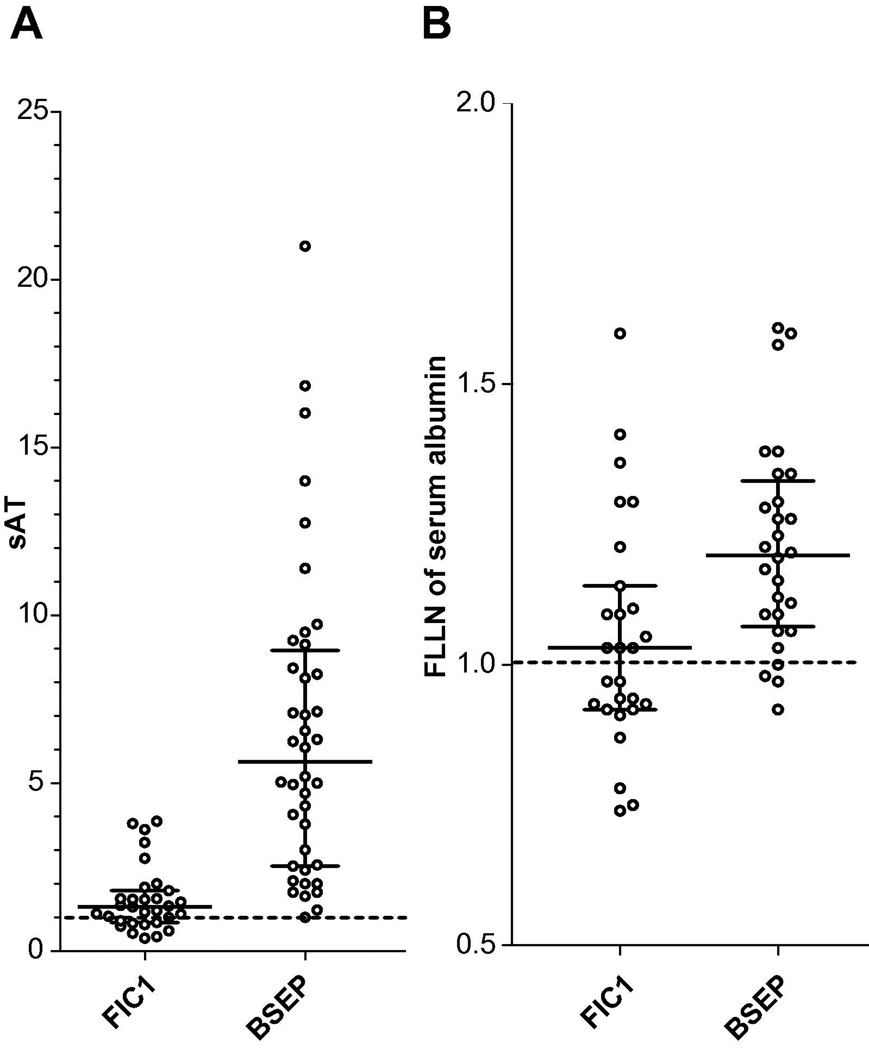
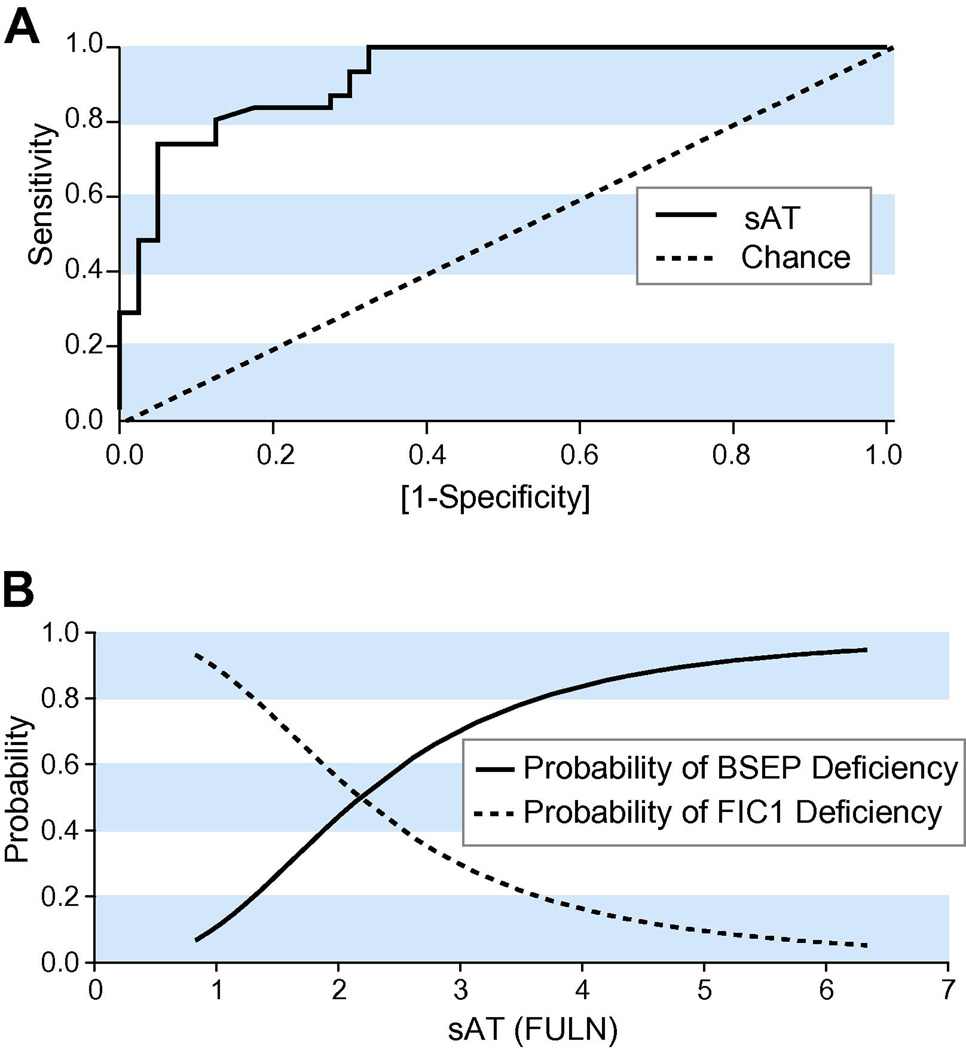
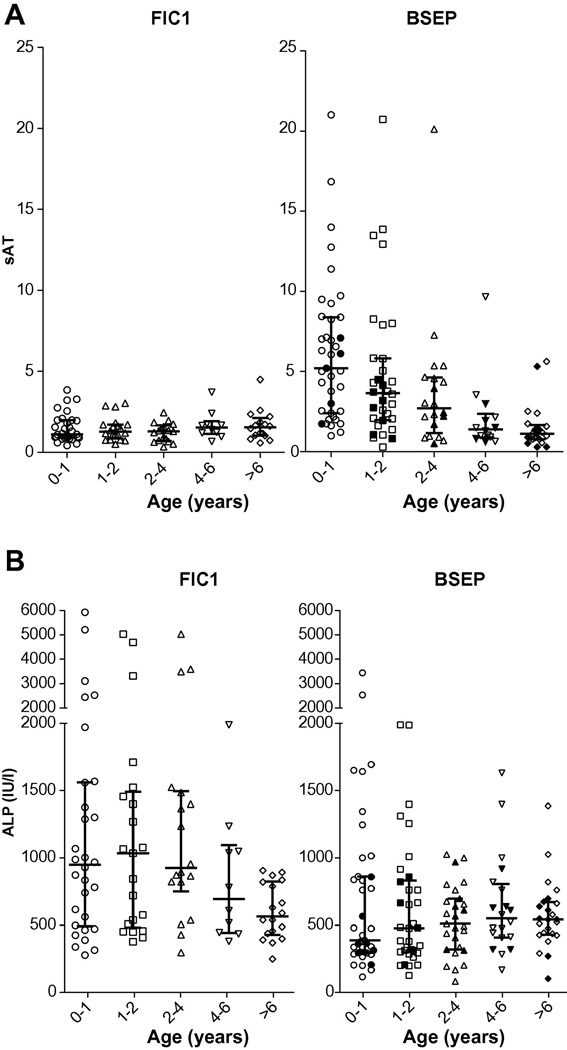
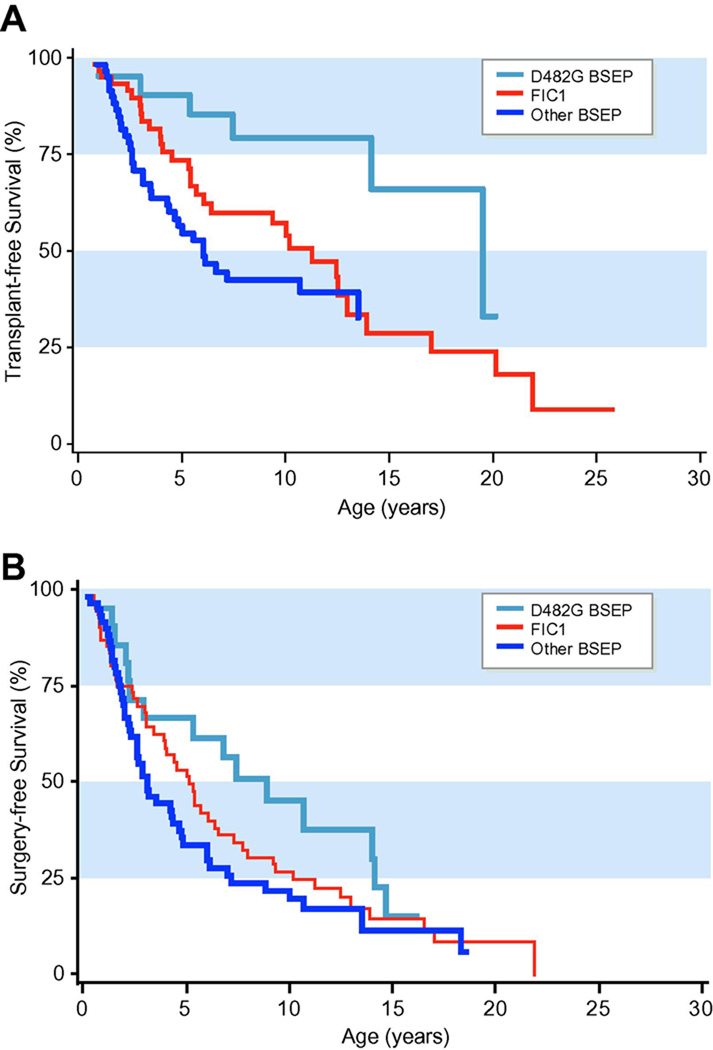
Results: At presentation, serum aminotransferase and bile salt levels were higher in BSEP patients; serum alkaline phosphatase values were higher, and serum albumin values were lower, in FIC1 patients. Elevated white blood cell counts, and giant or multinucleate cells at liver biopsy, were more common in BSEP patients. BSEP patients more often had gallstones and portal hypertension. Diarrhea, pancreatic disease, rickets, pneumonia, abnormal sweat tests, hearing impairment, and poor growth were more common in FIC1 patients. Among BSEP patients, the course of disease was less rapidly progressive in patients bearing the D482G mutation.
Conclusions: Severe forms of FIC1 and BSEP deficiency differed. BSEP patients manifested more severe hepatobiliary disease, while FIC1 patients showed greater evidence of extrahepatic disease.
Copyright 2010 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Conflicts of Interest: Dr. Steven Lobritto was on the Speakers’ Bureau of TAP Pharmaceuticals during the development of this manuscript, however, he is no longer on the speakers Bureau of TAP pharmaceuticals. No other conflicts of interest exist.
Figures
References
-
- Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–224. - PubMed
-
- Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. - PubMed
-
- Knisely AS, Bull L, Shneider BL. Low-γGT familial intrahepatic cholestasis. Gene Reviews: Clinical Genetic Information Resource. 2008
-
- Ujhazy P, Ortiz D, Misra S, Li S, Moseley J, Jones H, et al. Familial intrahepatic cholestasis 1: Studies of localization and function. Hepatology. 2001;34:768–775. - PubMed
-
- Paulusma CC, Folmer DE, Ho-Mok KS, de Waart DR, Hilarius PM, Verhoeven AJ, et al. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. 2008;47:268–278. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
