

Therapeutic targeting of mitochondrial superoxide in hypertension
- PMID: 20448215
- PMCID: PMC2901409
- DOI: 10.1161/CIRCRESAHA.109.214601
Therapeutic targeting of mitochondrial superoxide in hypertension
Abstract
Rationale: Superoxide (O2(-) ) has been implicated in the pathogenesis of many human diseases including hypertension; however, commonly used antioxidants have proven ineffective in clinical trials. It is possible that these agents are not adequately delivered to the subcellular sites of superoxide production.
Objective: Because the mitochondria are important sources of reactive oxygen species, we postulated that mitochondrial targeting of superoxide scavenging would have therapeutic benefit.
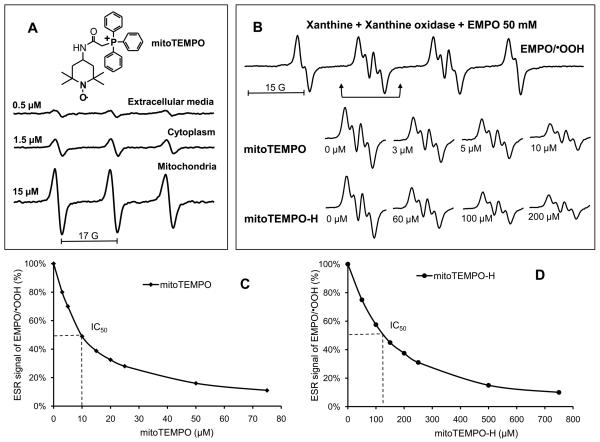
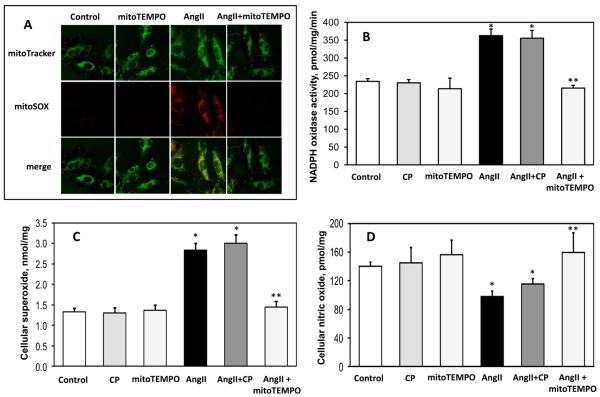
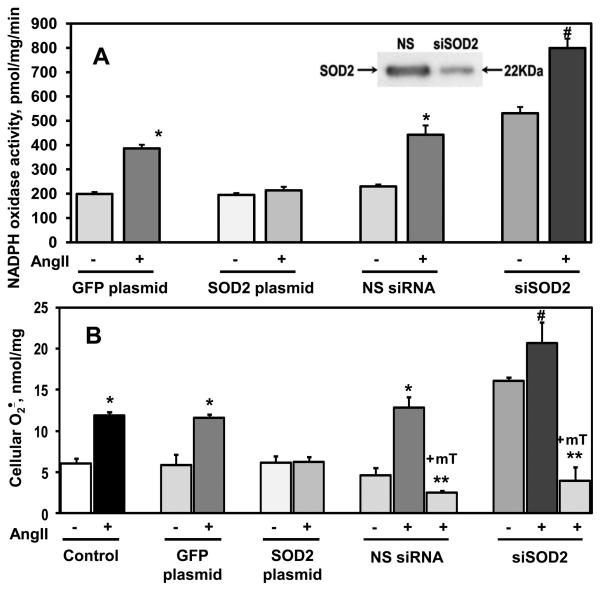
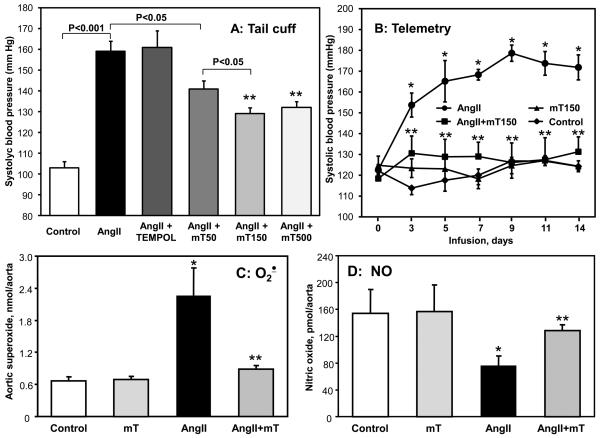
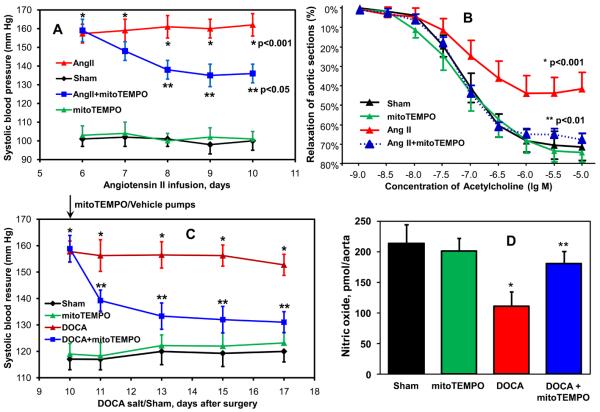
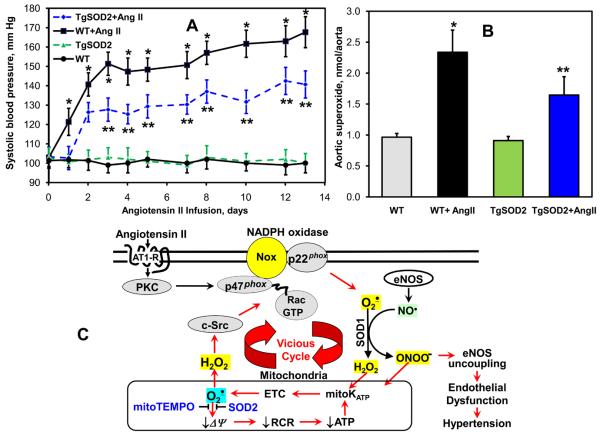
Methods and results: In this study, we found that the hormone angiotensin (Ang II) increased endothelial mitochondrial superoxide production. Treatment with the mitochondria-targeted antioxidant mitoTEMPO decreased mitochondrial O2(-), inhibited the total cellular O2(-), reduced cellular NADPH oxidase activity, and restored the level of bioavailable NO. These effects were mimicked by overexpressing the mitochondrial MnSOD (SOD2), whereas SOD2 depletion with small interfering RNA increased both basal and Ang II-stimulated cellular O2(-). Treatment of mice in vivo with mitoTEMPO attenuated hypertension when given at the onset of Ang II infusion and decreased blood pressure by 30 mm Hg following establishment of both Ang II-induced and DOCA salt hypertension, whereas a similar dose of nontargeted TEMPOL was not effective. In vivo, mitoTEMPO decreased vascular O2(-), increased vascular NO production and improved endothelial-dependent relaxation. Interestingly, transgenic mice overexpressing mitochondrial SOD2 demonstrated attenuated Ang II-induced hypertension and vascular oxidative stress similar to mice treated with mitoTEMPO.
Conclusions: These studies show that mitochondrial O2(-) is important for the development of hypertension and that antioxidant strategies specifically targeting this organelle could have therapeutic benefit in this and possibly other diseases.
Figures
Comment in
-
Resurrecting hope for antioxidant treatment of cardiovascular disease: focus on mitochondria.Circ Res. 2010 Jul 9;107(1):9-11. doi: 10.1161/CIRCRESAHA.110.223321. Circ Res. 2010. PMID: 20616335 Free PMC article. No abstract available.
References
-
- Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–473. - PubMed
-
- Puddu P, Puddu GM, Galletti L, Cravero E, Muscari A. Mitochondrial dysfunction as an initiating event in atherogenesis: a plausible hypothesis. Cardiology. 2005;103:137–141. - PubMed
-
- Rego AC, Oliveira CR. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem Res. 2003;28:1563–1574. - PubMed
-
- Wall JA, Wei J, Ly M, Belmont P, Martindale JJ, Tran D, Sun J, Chen WJ, Yu W, Oeller P, Briggs S, Gustafsson AB, Sayen MR, Gottlieb RA, Glembotski CC. Alterations in oxidative phosphorylation complex proteins in the hearts of transgenic mice that overexpress the p38 MAP kinase activator, MAP kinase kinase 6. Am J Physiol Heart Circ Physiol. 2006;291:H2462–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL06072808/HL/NHLBI NIH HHS/United States
- R01 HL094469/HL/NHLBI NIH HHS/United States
- P0-1 HL075209/HL/NHLBI NIH HHS/United States
- L058863/PHS HHS/United States
- R01 HL058863/HL/NHLBI NIH HHS/United States
- P01 HL058000/HL/NHLBI NIH HHS/United States
- P0-1 HL058000/HL/NHLBI NIH HHS/United States
- R01 HL124116/HL/NHLBI NIH HHS/United States
- R37 HL038206/HL/NHLBI NIH HHS/United States
- P01 HL075209/HL/NHLBI NIH HHS/United States
- R01 HL038206/HL/NHLBI NIH HHS/United States
- HL094469/HL/NHLBI NIH HHS/United States
- HL38206/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
