

Mechanisms of defibrillation
- PMID: 20450352
- PMCID: PMC3984906
- DOI: 10.1146/annurev-bioeng-070909-105305
Mechanisms of defibrillation
Abstract
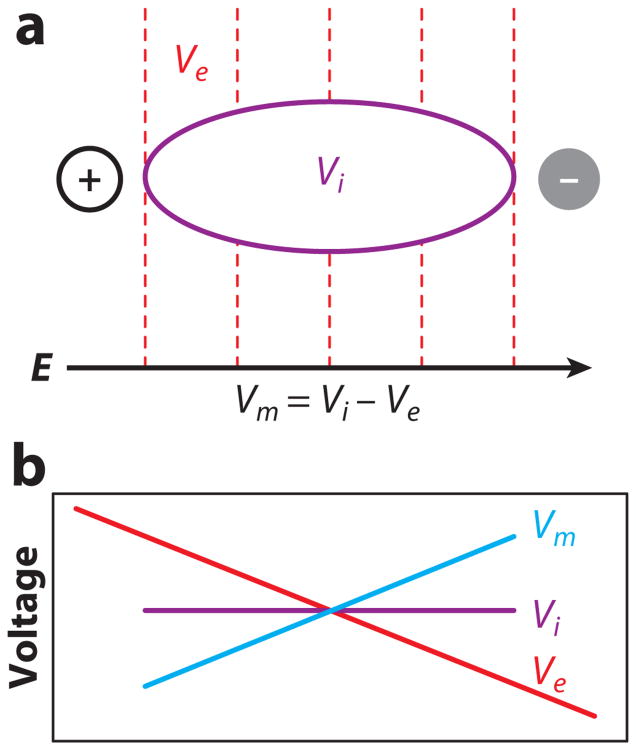
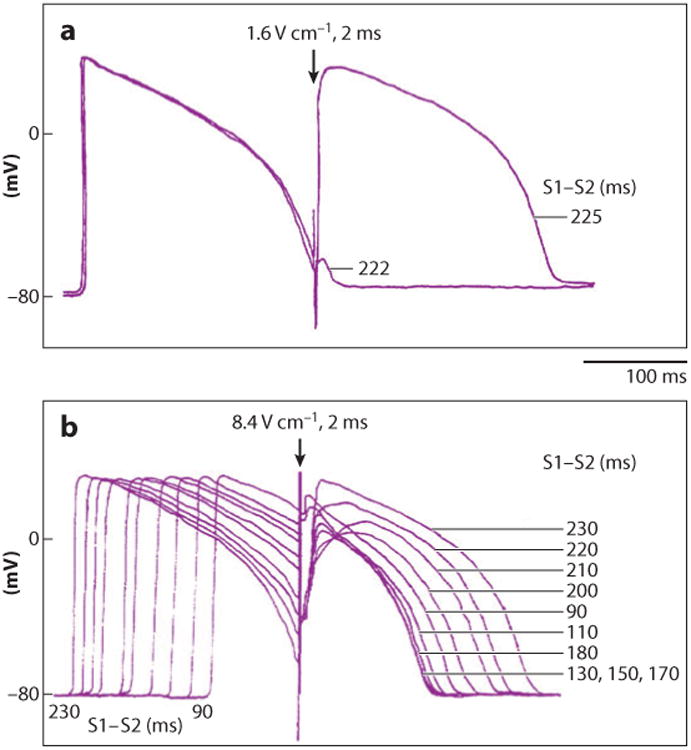
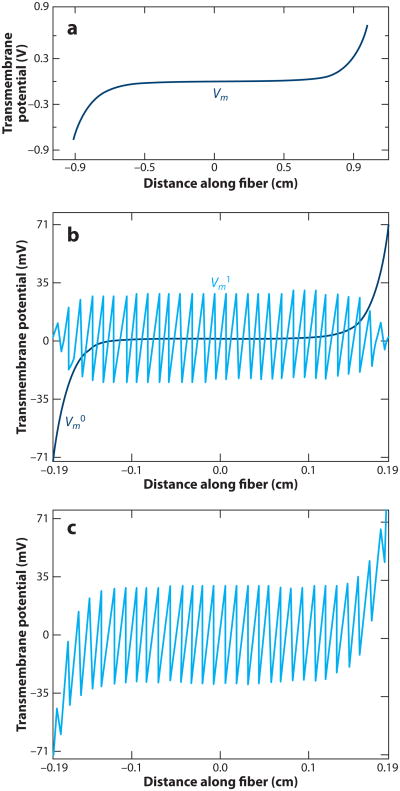
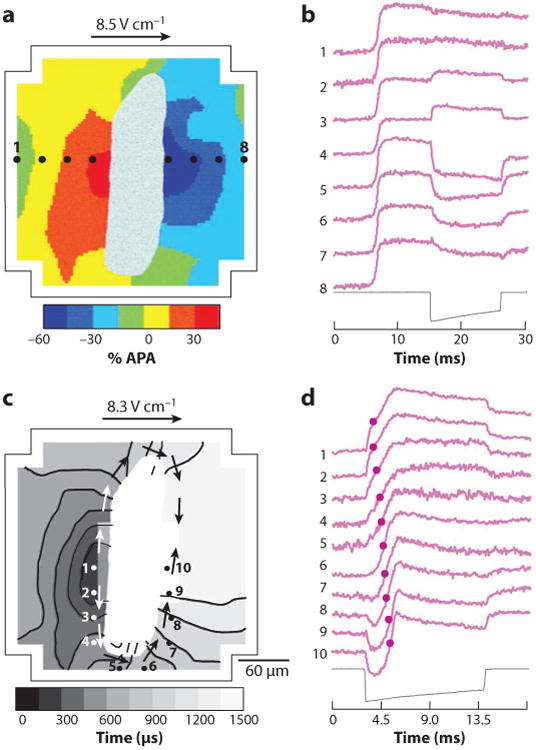
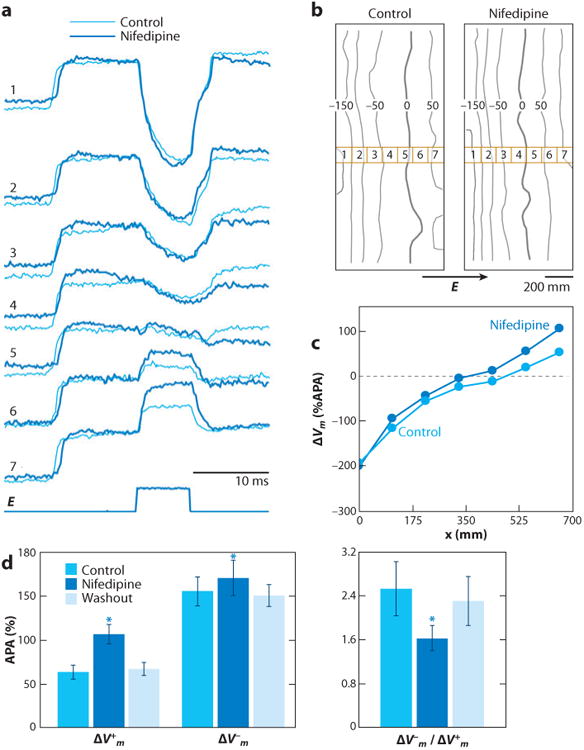
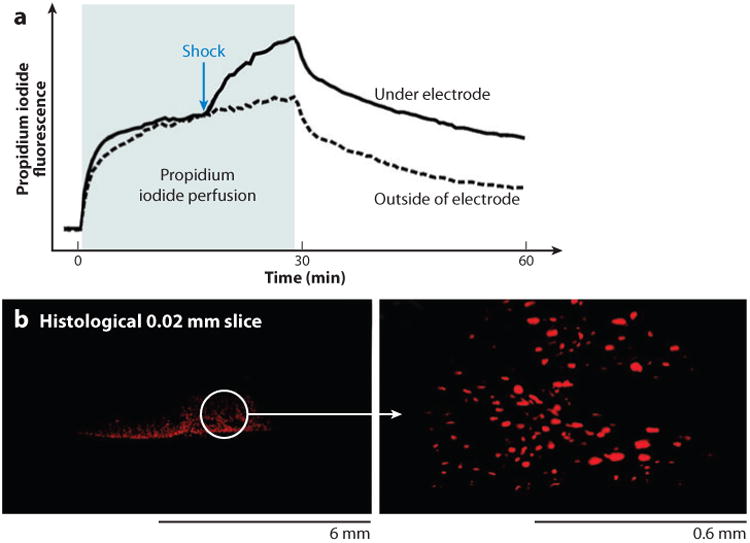
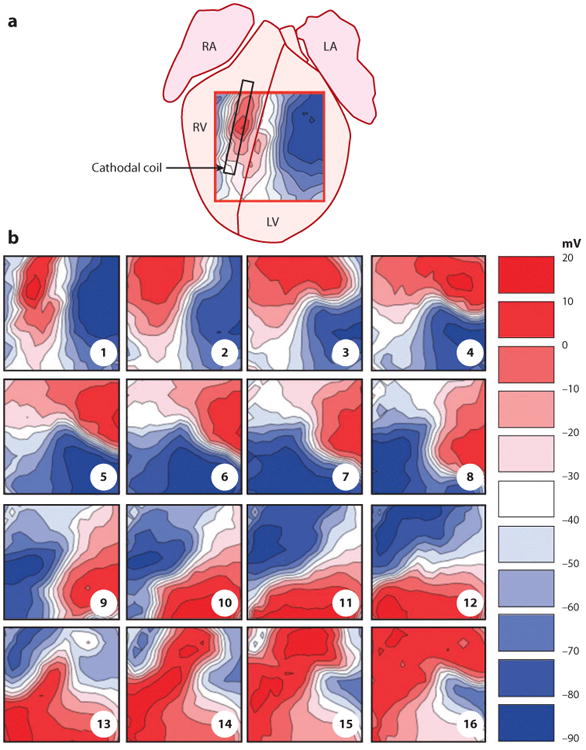
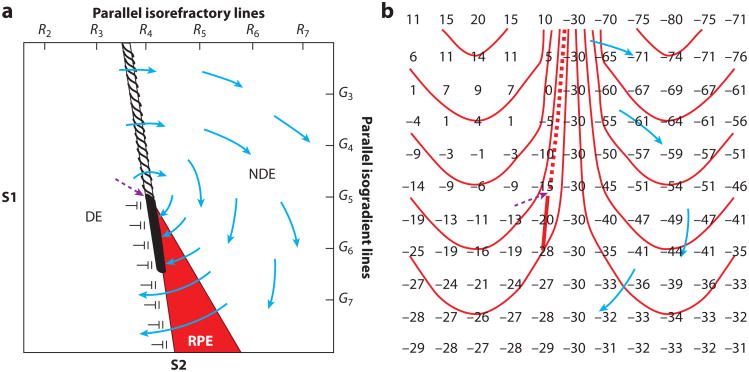
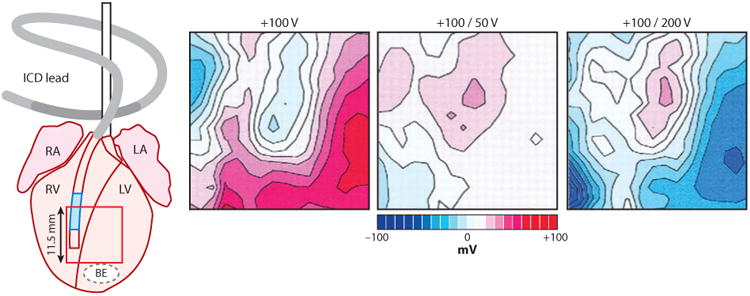
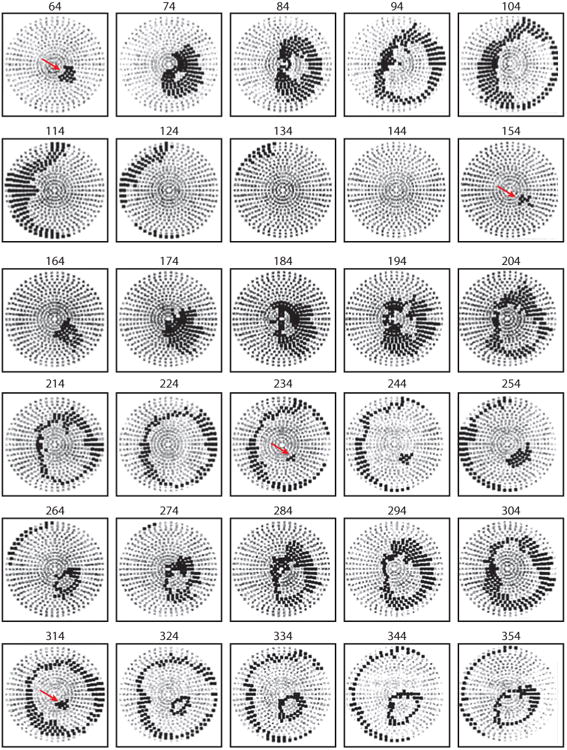
Electrical shock has been the one effective treatment for ventricular fibrillation for several decades. With the advancement of electrical and optical mapping techniques, histology, and computer modeling, the mechanisms responsible for defibrillation are now coming to light. In this review, we discuss recent work that demonstrates the various mechanisms responsible for defibrillation. On the cellular level, membrane depolarization and electroporation affect defibrillation outcome. Cell bundles and collagenous septae are secondary sources and cause virtual electrodes at sites far from shocking electrodes. On the whole-heart level, shock field gradient and critical points determine whether a shock is successful or whether reentry causes initiation and continuation of fibrillation.
Figures
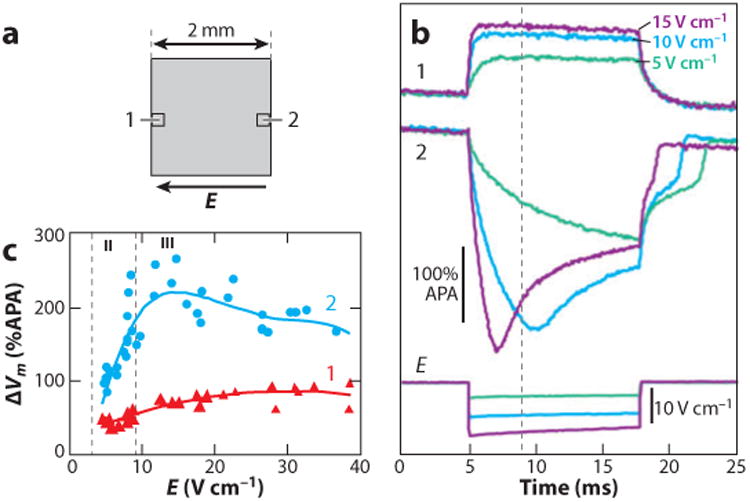
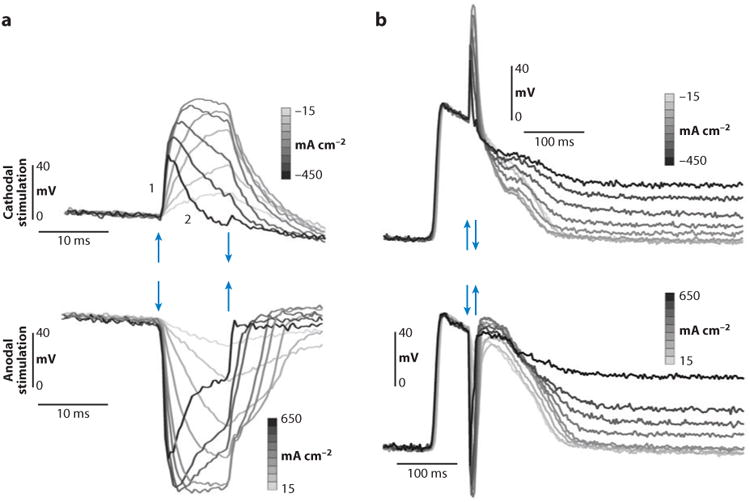
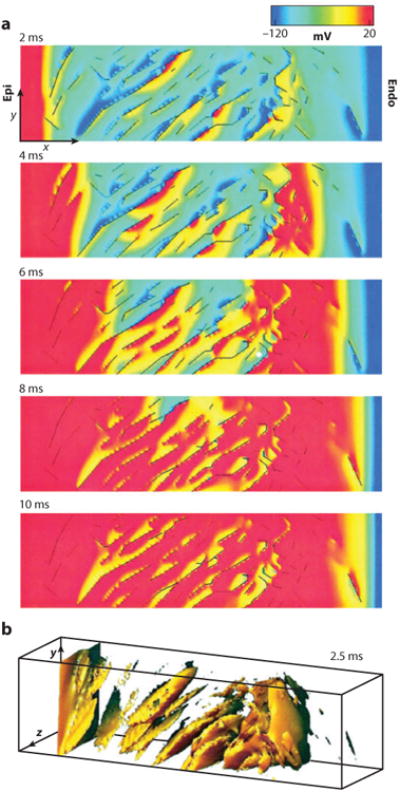
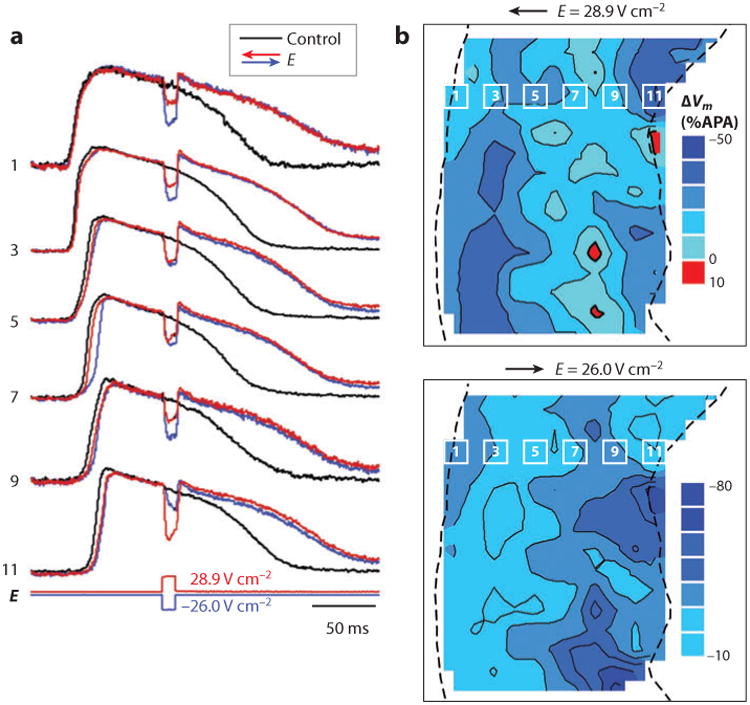
Similar articles
-
Imaging of Ventricular Fibrillation and Defibrillation: The Virtual Electrode Hypothesis.Adv Exp Med Biol. 2015;859:343-65. doi: 10.1007/978-3-319-17641-3_14. Adv Exp Med Biol. 2015. PMID: 26238060 Free PMC article. Review.
-
Mechanism of cardiac defibrillation in open-chest dogs with unipolar DC-coupled simultaneous activation and shock potential recordings.Circulation. 1990 Jul;82(1):244-60. doi: 10.1161/01.cir.82.1.244. Circulation. 1990. PMID: 2364513
-
Extension of refractoriness in a model of cardiac defibrillation.Pac Symp Biocomput. 1999:240-51. doi: 10.1142/9789814447300_0024. Pac Symp Biocomput. 1999. PMID: 10380201
-
Effects of high-frequency biphasic shocks on ventricular vulnerability and defibrillation outcomes through synchronized virtual electrode responses.PLoS One. 2020 May 1;15(5):e0232529. doi: 10.1371/journal.pone.0232529. eCollection 2020. PLoS One. 2020. PMID: 32357163 Free PMC article.
-
On the mechanism of ventricular defibrillation.Pacing Clin Electrophysiol. 1997 Feb;20(2 Pt 2):422-31. doi: 10.1111/j.1540-8159.1997.tb06201.x. Pacing Clin Electrophysiol. 1997. PMID: 9058846 Review.
Cited by
-
Experimental studies of spiral wave teleportation in a light sensitive Belousov-Zhabotinsky system.Chaos. 2024 Sep 1;34(9):093106. doi: 10.1063/5.0216649. Chaos. 2024. PMID: 39226479
-
Toward a More Efficient Implementation of Antifibrillation Pacing.PLoS One. 2016 Jul 8;11(7):e0158239. doi: 10.1371/journal.pone.0158239. eCollection 2016. PLoS One. 2016. PMID: 27391010 Free PMC article.
-
Diffuse, non-polar electropermeabilization and reduced propidium uptake distinguish the effect of nanosecond electric pulses.Biochim Biophys Acta. 2015 Oct;1848(10 Pt A):2118-25. doi: 10.1016/j.bbamem.2015.06.018. Epub 2015 Jun 22. Biochim Biophys Acta. 2015. PMID: 26112464 Free PMC article.
-
A real-time system for selectively sensing and pacing the His-bundle during sinus rhythm and ventricular fibrillation.Biomed Eng Online. 2020 Apr 10;19(1):19. doi: 10.1186/s12938-020-00763-6. Biomed Eng Online. 2020. PMID: 32276597 Free PMC article.
-
Excitation of murine cardiac myocytes by nanosecond pulsed electric field.J Cardiovasc Electrophysiol. 2019 Mar;30(3):392-401. doi: 10.1111/jce.13834. Epub 2019 Jan 17. J Cardiovasc Electrophysiol. 2019. PMID: 30582656 Free PMC article.
References
-
- Hoffa M, Ludwig C. Einige neue Versuche über Herzebewegung. Z Ration Med. 1850;9:107–44.
-
- Prevost JL, Battelli F. Sur quelques effets des décharges électriques sur le coeur des Mammifères. C R Acad Sci. 1899;129:1267–68.
-
- Beck CS, Pritchard WH, Feil HS. Ventricular fibrillation of long duration abolished by electric shock. JAMA. 1947;135:985–86. - PubMed
-
- Zoll PM, Linenthal AJ, Gibson W, Paul MH, Norman LR. Termination of ventricular fibrillation in man by externally applied electric countershock. N Engl J Med. 1956;254:727–32. - PubMed
-
- Zipes DP, Fischer J, King RM, Nicoll A, Jolly WW. Termination of ventricular fibrillation in dogs by depolarizing a critical amount of myocardium. Am J Cardiol. 1975;36:37–44. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
