

Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate
- PMID: 20451169
- PMCID: PMC2868995
- DOI: 10.1016/j.ajhg.2010.04.007
Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate
Abstract
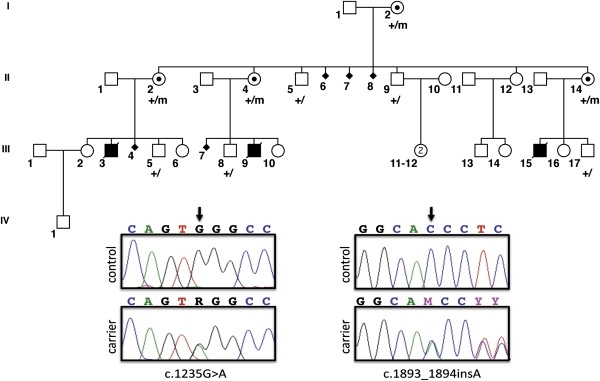
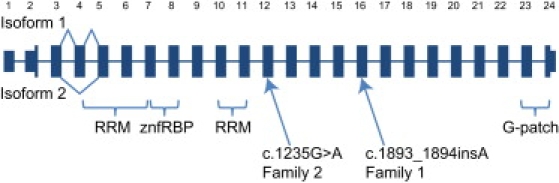
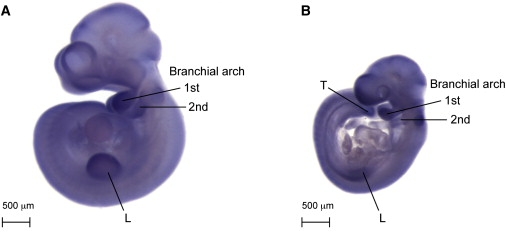
Micrognathia, glossoptosis, and cleft palate comprise one of the most common malformation sequences, Robin sequence. It is a component of the TARP syndrome, talipes equinovarus, atrial septal defect, Robin sequence, and persistent left superior vena cava. This disorder is X-linked and severe, with apparently 100% pre- or postnatal lethality in affected males. Here we characterize a second family with TARP syndrome, confirm linkage to Xp11.23-q13.3, perform massively parallel sequencing of X chromosome exons, filter the results via a number of criteria including the linkage region, use a unique algorithm to characterize sequence changes, and show that TARP syndrome is caused by mutations in the RBM10 gene, which encodes RNA binding motif 10. We further show that this previously uncharacterized gene is expressed in midgestation mouse embryos in the branchial arches and limbs, consistent with the human phenotype. We conclude that massively parallel sequencing is useful to characterize large candidate linkage intervals and that it can be used successfully to allow identification of disease-causing gene mutations.
Copyright (c) 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Gorlin R.J., Cervenka J., Anderson R.C., Sauk J.J., Bevis W.D. Robin's syndrome. A probably X-linked recessive subvariety exhibiting persistence of left superior vena cava and atrial septal defect. Am. J. Dis. Child. 1970;119:176–178. - PubMed
-
- Kurpinski K.T., Magyari P.A., Gorlin R.J., Ng D., Biesecker L.G. Designation of the TARP syndrome and linkage to Xp11.23-q13.3 without samples from affected patients. Am. J. Med. Genet. A. 2003;120A:1–4. - PubMed
-
- Johnston J.J., Olivos-Glander I., Killoran C., Elson E., Turner J.T., Peters K.F., Abbott M.H., Aughton D.J., Aylsworth A.S., Bamshad M.J. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: Robust phenotype prediction from the type and position of GLI3 mutations. Am. J. Hum. Genet. 2005;76:609–622. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
