

Endothelin-1 (ET-1) increases the expression of remodeling genes in vascular smooth muscle through linked calcium and cAMP pathways: role of a phospholipase A(2)(cPLA(2))/cyclooxygenase-2 (COX-2)/prostacyclin receptor-dependent autocrine loop
- PMID: 20452970
- PMCID: PMC2923981
- DOI: 10.1074/jbc.M110.139485
Endothelin-1 (ET-1) increases the expression of remodeling genes in vascular smooth muscle through linked calcium and cAMP pathways: role of a phospholipase A(2)(cPLA(2))/cyclooxygenase-2 (COX-2)/prostacyclin receptor-dependent autocrine loop
Abstract
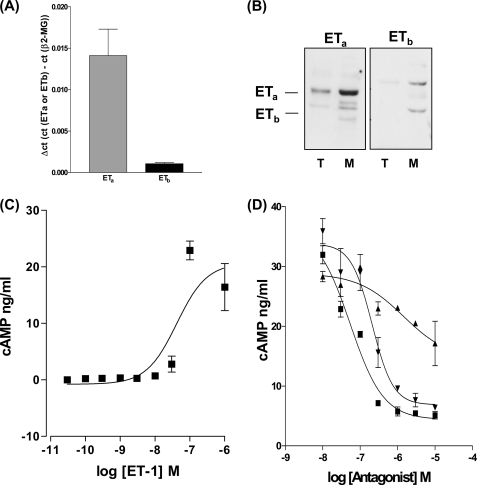
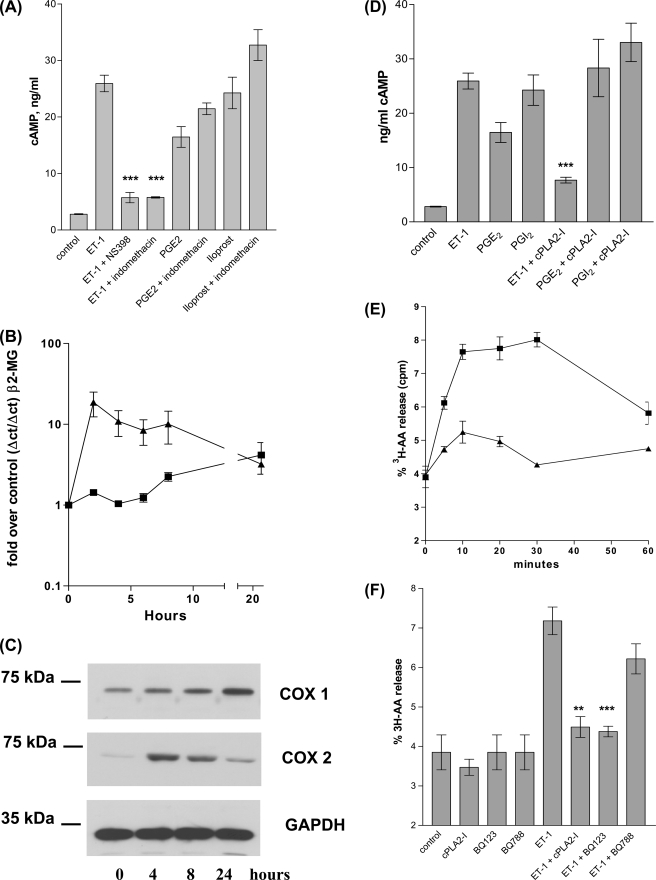
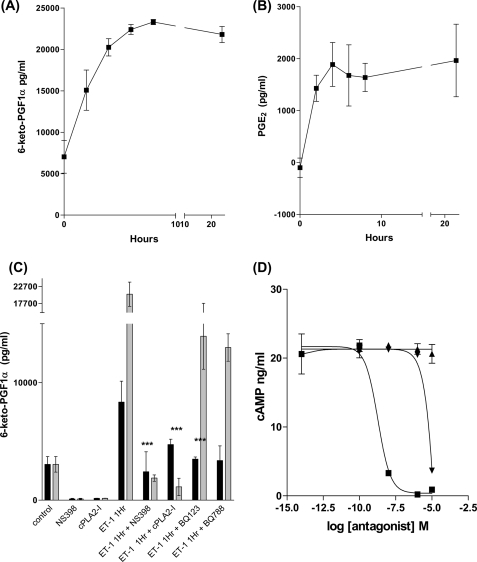
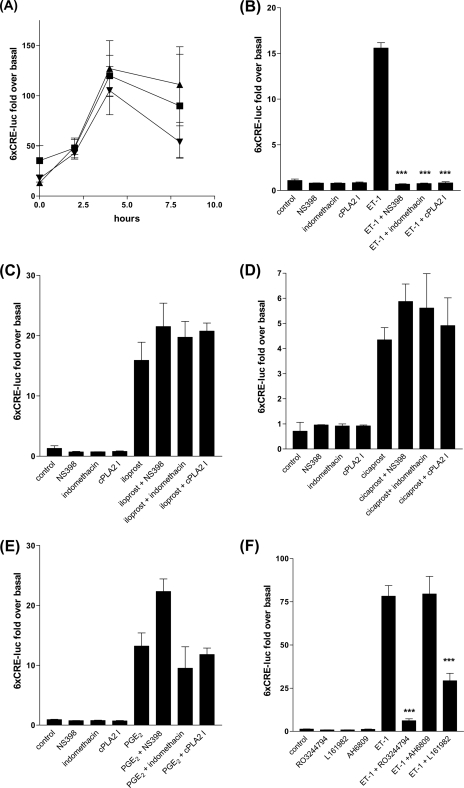
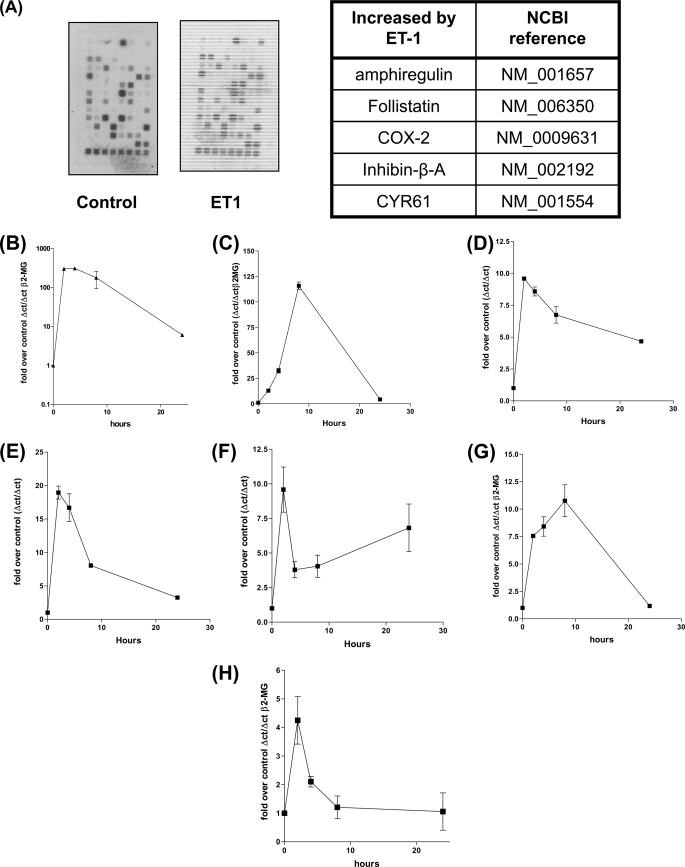
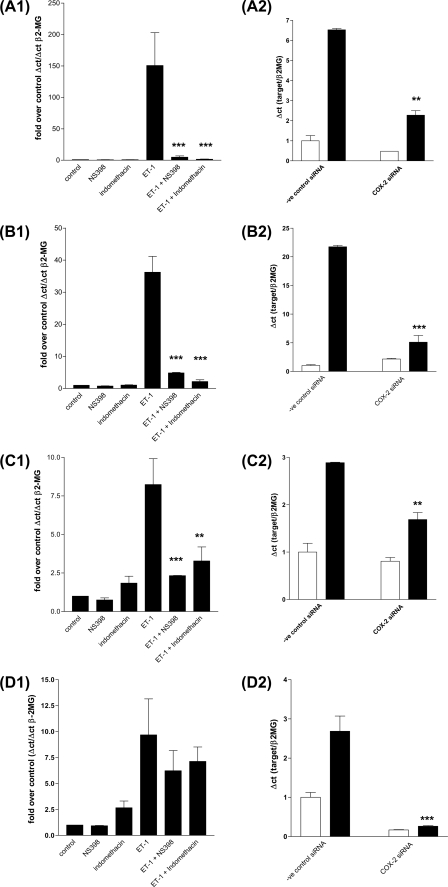
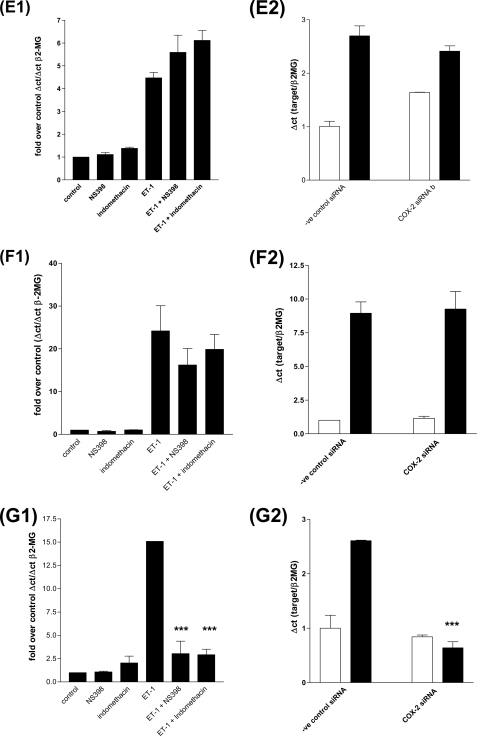
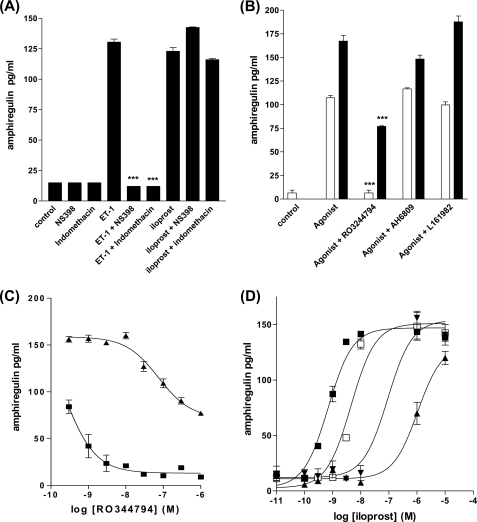
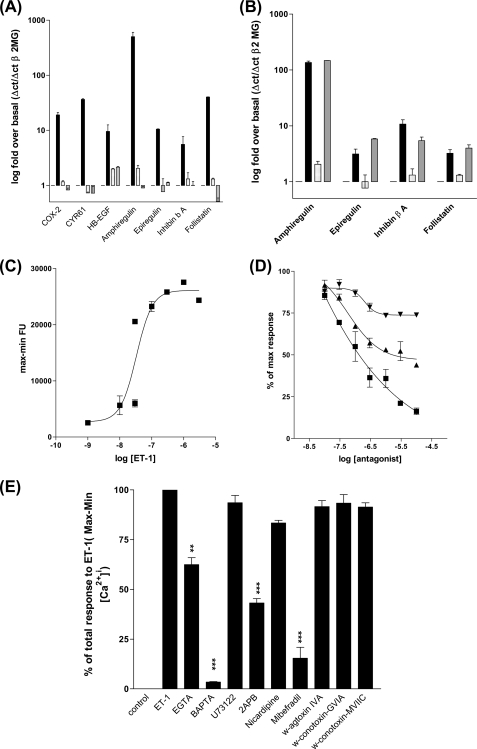
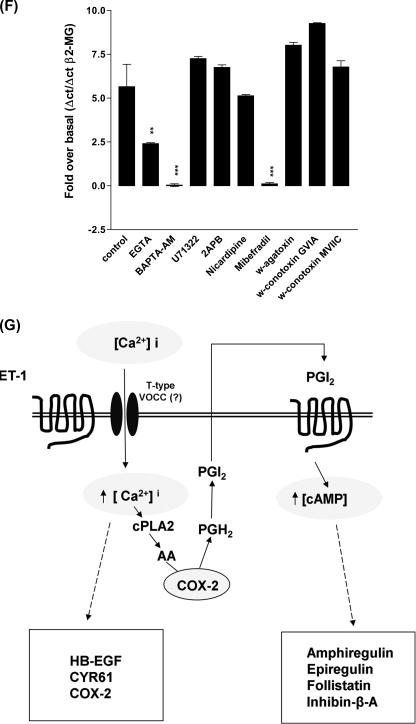
Several important genes that are involved in inflammation and tissue remodeling are switched on by virtue of CRE response elements in their promoters. The upstream signaling mechanisms that inflammatory mediators use to activate cAMP response elements (CREs) are poorly understood. Endothelin (ET) is an important vasoactive mediator that plays roles in inflammation, vascular remodeling, angiogenesis, and carcinogenesis by activating 7 transmembrane G protein-coupled receptors (GPCR). Here we characterized the mechanisms ET-1 uses to regulate CRE-dependent remodeling genes in pulmonary vascular smooth muscle cells. These studies revealed activation pathways involving a cyclooxygenase-2 (COX-2)/prostacyclin receptor (IP receptor) autocrine loop and an interlinked calcium-dependent pathway. We found that ET-1 activated several CRE response genes in vascular smooth muscle cells, particularly COX-2, amphiregulin, follistatin, inhibin-beta-A, and CYR61. ET-1 also activated two other genes epiregulin and HB-EGF. Amphiregulin, follistatin, and inhibin-beta-A and epiregulin were activated by an autocrine loop involving cPLA2, arachidonic acid release, COX-2-dependent PGI(2) synthesis, and IP receptor-linked elevation of cAMP leading to CRE transcription activation. In contrast COX-2, CYR61, and HB-EGF transcription were regulated in a calcium-dependent, COX-2 independent, manner. Observations with IP receptor antagonists and COX-2 inhibitors were confirmed with IP receptor or COX-2-specific small interfering RNAs. ET-1 increases in intracellular calcium and gene transcription were dependent upon ET(a) activation and calcium influx through T type voltage-dependent calcium channels. These studies give important insights into the upstream signaling mechanisms used by G protein-coupled receptor-linked mediators such as ET-1, to activate CRE response genes involved in angiogenesis, vascular remodeling, inflammation, and carcinogenesis.
Figures

Similar articles
-
Cyclooxygenase-2 induction by bradykinin in human pulmonary artery smooth muscle cells is mediated by the cyclic AMP response element through a novel autocrine loop involving endogenous prostaglandin E2, E-prostanoid 2 (EP2), and EP4 receptors.J Biol Chem. 2003 Dec 12;278(50):49954-64. doi: 10.1074/jbc.M307964200. Epub 2003 Sep 29. J Biol Chem. 2003. PMID: 14517215
-
Gbetagamma-mediated prostacyclin production and cAMP-dependent protein kinase activation by endothelin-1 promotes vascular smooth muscle cell hypertrophy through inhibition of glycogen synthase kinase-3.J Biol Chem. 2007 Jul 6;282(27):19518-25. doi: 10.1074/jbc.M702655200. Epub 2007 May 19. J Biol Chem. 2007. PMID: 17513863
-
Endothelin-1 activates p38 mitogen-activated protein kinase and cytosolic phospholipase A2 in cat iris sphincter smooth muscle cells.Biochem J. 1999 Aug 15;342 ( Pt 1)(Pt 1):87-96. Biochem J. 1999. PMID: 10432304 Free PMC article.
-
Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn.Pharmacol Rev. 2012 Jul;64(3):540-82. doi: 10.1124/pr.111.004770. Epub 2012 Jun 7. Pharmacol Rev. 2012. PMID: 22679221 Free PMC article. Review.
-
New insights into the intracellular mechanisms by which PGI2 analogues elicit vascular relaxation: cyclic AMP-independent, Gs-protein mediated-activation of MaxiK channel.Curr Med Chem Cardiovasc Hematol Agents. 2004 Jul;2(3):257-65. doi: 10.2174/1568016043356273. Curr Med Chem Cardiovasc Hematol Agents. 2004. PMID: 15320791 Review.
Cited by
-
An autophagic deficit in the uterine vessel microenvironment provokes hyperpermeability through deregulated VEGFA, NOS1, and CTNNB1.Autophagy. 2021 Jul;17(7):1649-1666. doi: 10.1080/15548627.2020.1778292. Epub 2020 Jun 17. Autophagy. 2021. PMID: 32579471 Free PMC article.
-
PDGF-BB regulates the pulmonary vascular tone: impact of prostaglandins, calcium, MAPK- and PI3K/AKT/mTOR signalling and actin polymerisation in pulmonary veins of guinea pigs.Respir Res. 2018 Jun 19;19(1):120. doi: 10.1186/s12931-018-0829-5. Respir Res. 2018. PMID: 29921306 Free PMC article.
-
Static Stretch Increases the Pro-Inflammatory Response of Rat Type 2 Alveolar Epithelial Cells to Dynamic Stretch.Front Physiol. 2022 Apr 11;13:838834. doi: 10.3389/fphys.2022.838834. eCollection 2022. Front Physiol. 2022. PMID: 35480037 Free PMC article.
-
Intra-city Differences in Cardiac Expression of Inflammatory Genes and Inflammasomes in Young Urbanites: A Pilot Study.J Toxicol Pathol. 2012 Jun;25(2):163-73. doi: 10.1293/tox.25.163. J Toxicol Pathol. 2012. PMID: 22907983 Free PMC article.
-
Deletion of T-type calcium channels Cav3.1 or Cav3.2 attenuates endothelial dysfunction in aging mice.Pflugers Arch. 2018 Feb;470(2):355-365. doi: 10.1007/s00424-017-2068-x. Epub 2017 Oct 29. Pflugers Arch. 2018. PMID: 29082441
References
-
- Yanagisawa M., Kurihara H., Kimura S., Tomobe Y., Kobayashi M., Mitsui Y., Yazaki Y., Goto K., Masaki T. (1988) Nature 332, 411–415 - PubMed
-
- Woods M., Mitchell J. A., Wood E. G., Barker S., Walcot N. R., Rees G. M., Warner T. D. (1999) Mol. Pharmacol. 55, 902–909 - PubMed
-
- Suzuki T., Kumazaki T., Mitsui Y. (1993) Biochem. Biophys. Res. Commun. 191, 823–830 - PubMed
-
- Markewitz B. A., Kohan D. E., Michael J. R. (1995) Am. J. Physiol. Lung Cell. Mol. Physiol. 268, L192–L200 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
