

Expression of a Src family kinase in chronic myelogenous leukemia cells induces resistance to imatinib in a kinase-dependent manner
- PMID: 20452982
- PMCID: PMC2898387
- DOI: 10.1074/jbc.M109.090043
Expression of a Src family kinase in chronic myelogenous leukemia cells induces resistance to imatinib in a kinase-dependent manner
Abstract
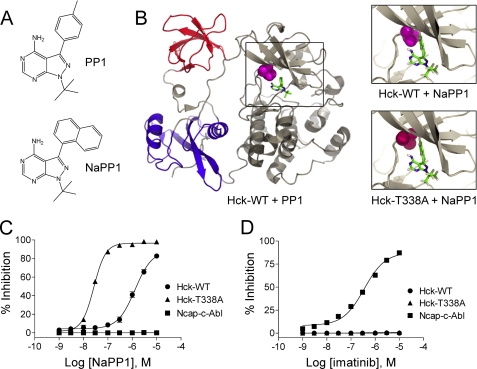
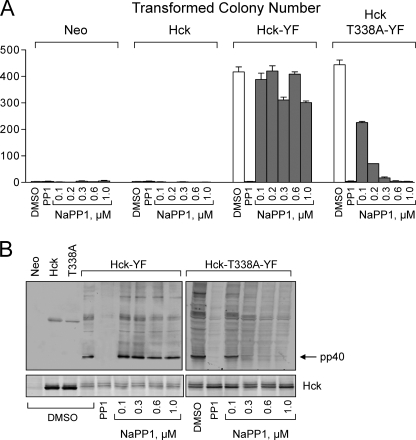
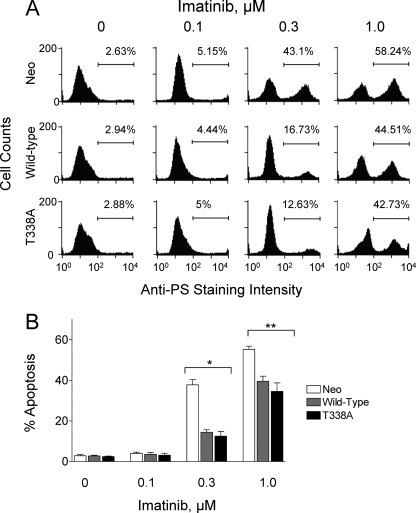
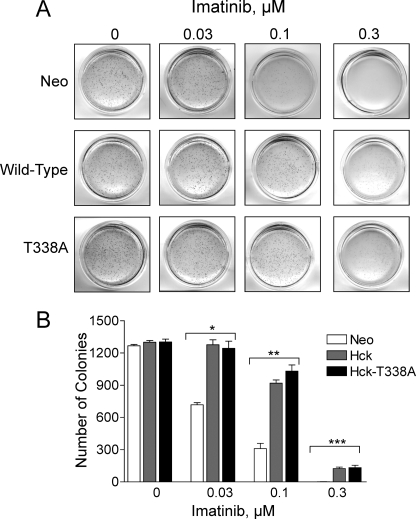
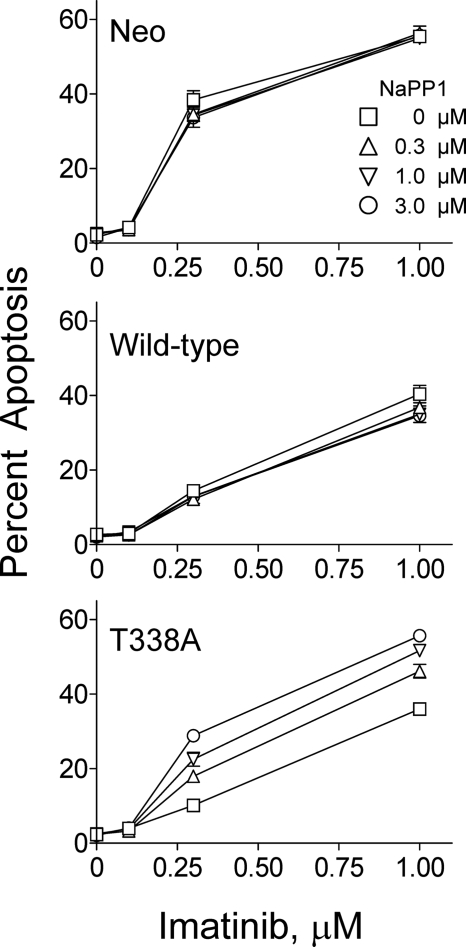
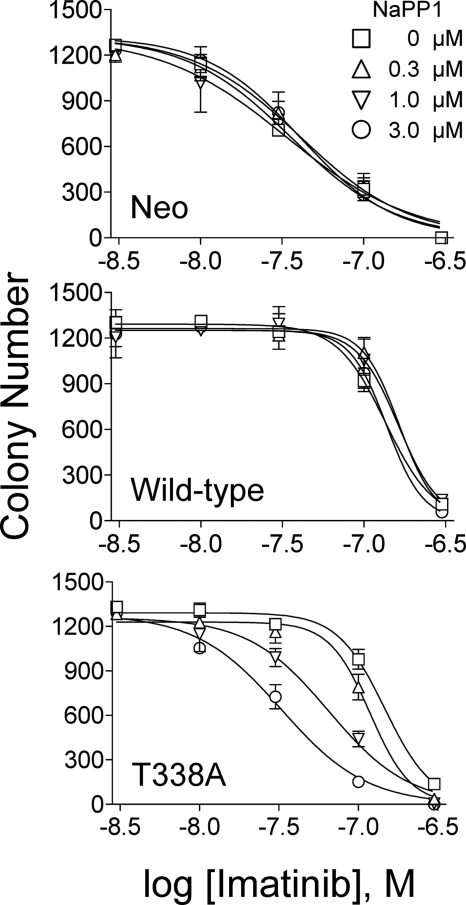
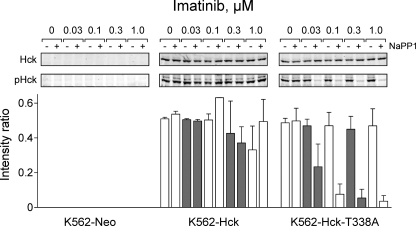
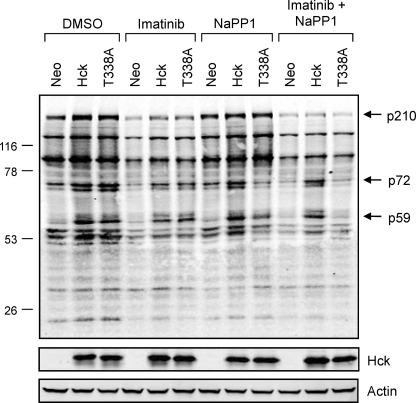
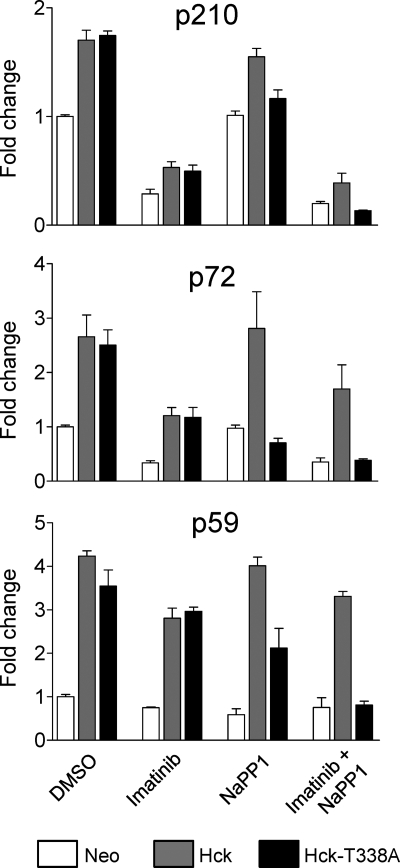
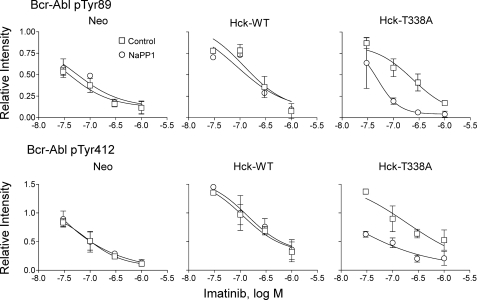
The Bcr-Abl kinase inhibitor imatinib is remarkably effective in chronic myelogenous leukemia (CML), although drug resistance is an emerging problem. Myeloid Src family kinases such as Hck and Lyn are often overexpressed in imatinib-resistant CML cells that lack Bcr-Abl mutations. Here we tested whether Hck overexpression is sufficient to induce imatinib resistance using both wild-type Hck and a mutant (Hck-T338A) that is uniquely sensitive to the pyrazolo-pyrimidine inhibitor, NaPP1. Expression of either kinase in K562 CML cells caused resistance to imatinib-induced apoptosis and inhibition of soft-agar colony formation. Treatment with NaPP1 restored sensitivity to imatinib in cells expressing T338A but not wild-type Hck, demonstrating that resistance requires Hck kinase activity. NaPP1 also reduced Hck-mediated phosphorylation of Bcr-Abl at sites that may affect imatinib sensitivity exclusively in cells expressing Hck-T338A. These data show that elevated Src family kinase activity is sufficient to induce imatinib resistance through a mechanism that may involve phosphorylation of Bcr-Abl.
Figures
Similar articles
-
Bortezomib and flavopiridol interact synergistically to induce apoptosis in chronic myeloid leukemia cells resistant to imatinib mesylate through both Bcr/Abl-dependent and -independent mechanisms.Blood. 2004 Jul 15;104(2):509-18. doi: 10.1182/blood-2003-12-4121. Epub 2004 Mar 23. Blood. 2004. PMID: 15039284
-
Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib-resistant chronic myelogenous leukemia cells.Blood. 2008 Apr 1;111(7):3821-9. doi: 10.1182/blood-2007-08-109330. Epub 2008 Jan 30. Blood. 2008. PMID: 18235045 Free PMC article.
-
An inhibitor-resistant mutant of Hck protects CML cells against the antiproliferative and apoptotic effects of the broad-spectrum Src family kinase inhibitor A-419259.Oncogene. 2008 Nov 27;27(56):7055-69. doi: 10.1038/onc.2008.330. Epub 2008 Sep 15. Oncogene. 2008. PMID: 18794796 Free PMC article.
-
Overcoming kinase resistance in chronic myeloid leukemia.Int J Biochem Cell Biol. 2008;40(3):334-43. doi: 10.1016/j.biocel.2007.10.001. Int J Biochem Cell Biol. 2008. PMID: 18401881 Review.
-
Targeting the kinase activity of the BCR-ABL fusion protein in patients with chronic myeloid leukemia.Curr Mol Med. 2005 Nov;5(7):615-23. doi: 10.2174/156652405774641115. Curr Mol Med. 2005. PMID: 16305488 Review.
Cited by
-
Current concepts in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia.Front Oncol. 2014 Mar 25;4:54. doi: 10.3389/fonc.2014.00054. eCollection 2014. Front Oncol. 2014. PMID: 24724051 Free PMC article. Review.
-
Hematopoietic cell kinase (HCK) as a therapeutic target in immune and cancer cells.Oncotarget. 2015 Jun 30;6(18):15752-71. doi: 10.18632/oncotarget.4199. Oncotarget. 2015. PMID: 26087188 Free PMC article. Review.
-
Overexpression of P-glycoprotein induces acquired resistance to imatinib in chronic myelogenous leukemia cells.Chin J Cancer. 2012 Feb;31(2):110-8. doi: 10.5732/cjc.011.10327. Epub 2011 Nov 18. Chin J Cancer. 2012. PMID: 22098951 Free PMC article.
-
Role of STAT3 in Transformation and Drug Resistance in CML.Front Oncol. 2012 Apr 10;2:30. doi: 10.3389/fonc.2012.00030. eCollection 2012. Front Oncol. 2012. PMID: 22649784 Free PMC article.
-
Dual-inhibitors of STAT5 and STAT3: studies from molecular docking and molecular dynamics simulations.J Mol Model. 2014 Aug;20(8):2399. doi: 10.1007/s00894-014-2399-x. Epub 2014 Aug 7. J Mol Model. 2014. PMID: 25098340
References
-
- Deininger M. W., Goldman J. M., Melo J. V. (2000) Blood 96, 3343–3356 - PubMed
-
- Groffen J., Stephenson J. R., Heisterkamp N., de Klein A., Bartram C. R., Grosveld G. (1984) Cell 36, 93–99 - PubMed
-
- Shtivelman E., Lifshitz B., Gale R. P., Canaani E. (1985) Nature 315, 550–554 - PubMed
-
- Raitano A. B., Whang Y. E., Sawyers C. L. (1997) Biochim. Biophys. Acta 1333, F201–F216 - PubMed
-
- Hantschel O., Superti-Furga G. (2004) Nat. Rev. Mol. Cell Biol. 5, 33–44 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
