

Laforin, the most common protein mutated in Lafora disease, regulates autophagy
- PMID: 20453062
- PMCID: PMC2893813
- DOI: 10.1093/hmg/ddq190
Laforin, the most common protein mutated in Lafora disease, regulates autophagy
Abstract
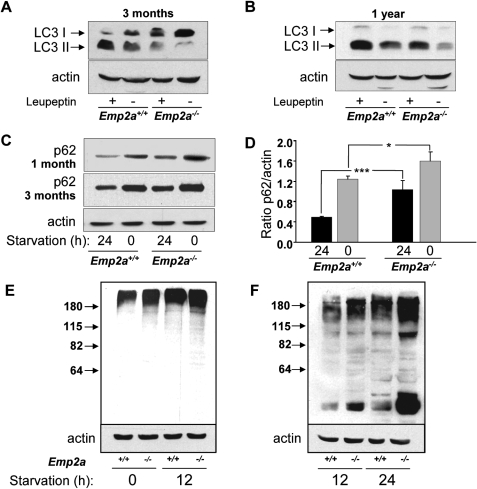
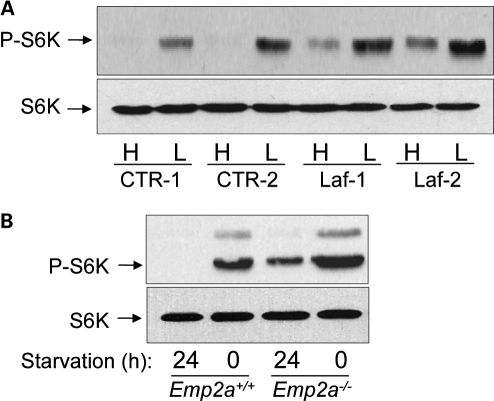
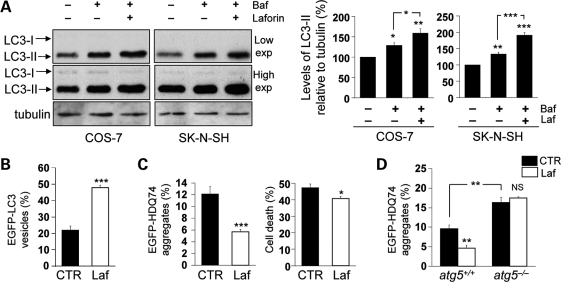
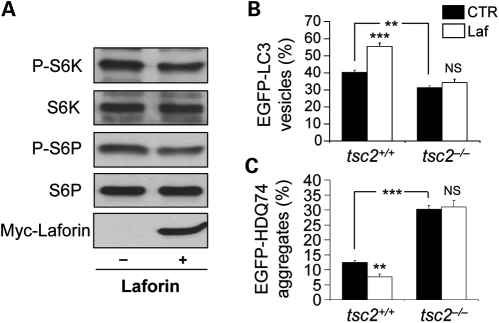
Lafora disease (LD) is an autosomal recessive, progressive myoclonus epilepsy, which is characterized by the accumulation of polyglucosan inclusion bodies, called Lafora bodies, in the cytoplasm of cells in the central nervous system and in many other organs. However, it is unclear at the moment whether Lafora bodies are the cause of the disease, or whether they are secondary consequences of a primary metabolic alteration. Here we describe that the major genetic lesion that causes LD, loss-of-function of the protein laforin, impairs autophagy. This phenomenon is confirmed in cell lines from human patients, mouse embryonic fibroblasts from laforin knockout mice and in tissues from such mice. Conversely, laforin expression stimulates autophagy. Laforin regulates autophagy via the mammalian target of rapamycin kinase-dependent pathway. The changes in autophagy mediated by laforin regulate the accumulation of diverse autophagy substrates and would be predicted to impact on the Lafora body accumulation and the cell stress seen in this disease that may eventually contribute to cell death.
Figures



References
-
- Lafora G.R., Glueck B. Beitrag zur Histopathologie der myoklonischen Epilepsie. Z. Gesammte Neurol. Psychiatr. 1911;6:1–14. doi:10.1007/BF02863929. - DOI
-
- Van Hoof F., Hageman-Bal M. Progressive familial myoclonic epilepsy with Lafora bodies. Electron microscopic and histochemical study of a cerebral biopsy. Acta Neuropathol. 1967;7:315–336. - PubMed
-
- Yokoi S., Nakayama H., Negishi T. Biochemical studies on tissues from a patient with Lafora disease. Clin. Chim. Acta. 1975;62:415–423. - PubMed
-
- Ganesh S., Delgado-Escueta A.V., Suzuki T., Francheschetti S., Riggio C., Avanzini G., Rabinowicz A., Bohlega S., Bailey J., Alonso M.E., et al. Genotype-phenotype correlations for EPM2A mutations in Lafora's progressive myoclonus epilepsy: exon 1 mutations associate with an early-onset cognitive deficit subphenotype. Hum. Mol. Genet. 2002;11:1263–1271. doi:10.1093/hmg/11.11.1263. - DOI - PubMed
-
- Sinha S., Satishchandra P., Gayathri N., Yasha T.C., Shankar S.K. Progressive myoclonic epilepsy: a clinical, electrophysiological and pathological study from South India. J. Neurol. Sci. 2007;252:16–23. doi:10.1016/j.jns.2006.09.021. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
