

Dominant negative p38 mitogen-activated protein kinase expression inhibits NF-kappaB activation in AR42J cells
- PMID: 20453549
- PMCID: PMC2899148
- DOI: 10.1159/000290656
Dominant negative p38 mitogen-activated protein kinase expression inhibits NF-kappaB activation in AR42J cells
Abstract
Background: The role of the p38 mitogen-activated protein (MAP) kinase in acute pancreatitis pathogenesis is controversial. We hypothesize that p38 plays a role in regulating NF-kappaB activation in exocrine pancreatic cells.
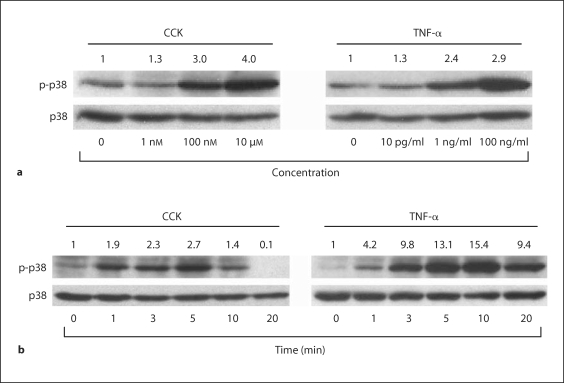
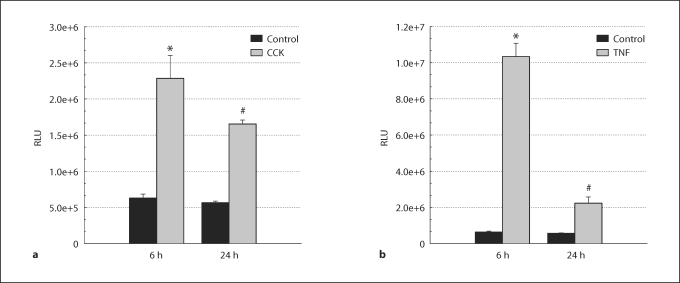
Methods: AR42J cells incorporating an NF-kappaB-responsive luciferase reporter, with and without adenoviral transduction of DNp38, were stimulated with cholecystokinin (CCK) or tumor necrosis factor-alpha (TNF-alpha) prior to measuring NF-kappaB activation.
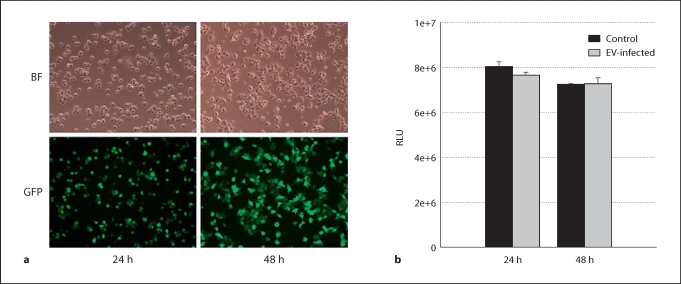
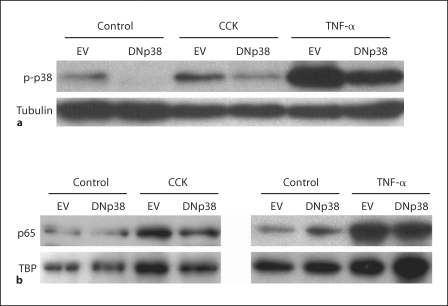
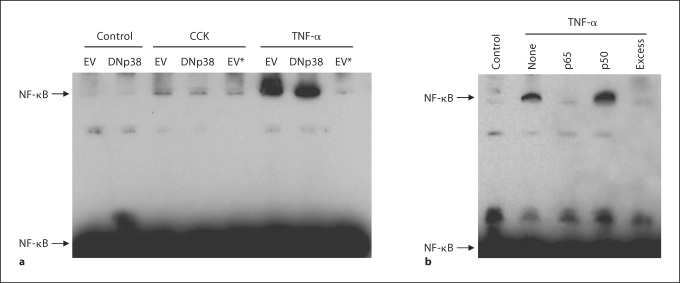
Results: CCK- or TNF-alpha-stimulated NF-kappaB-dependent gene transcription (luciferase assay) was substantially subdued by DNp38 expression. These findings were confirmed by electrophoretic mobility shift assay. Nuclear translocation of the p65 NF-kappaB subunit following agonist stimulation was evident (supershift). Characterization studies showed excellent adenoviral infection efficiency and cell viability in our AR42J cell model. Agonist-stimulated dose- and time-dependent p38 activation, with inhibition by DNp38 expression, was also confirmed.
Conclusion: The p38 MAP kinase regulates NF-kappaB pathway activation in exocrine pancreatic cells, and thus potentially plays a role in the mechanism of acute pancreatitis pathogenesis..
Copyright 2010 S. Karger AG, Basel.
Figures

References
-
- Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg. 1998;175:76–83. - PubMed
-
- Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD. NF-κB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology. 2002;122:448–457. - PubMed
-
- Han B, Ji B, Logsdon CD. CCK independently activates intracellular trypsinogen and NF-κB in rat pancreatic acinar cells. Am J Physiol. 2001;280:C465–C472. - PubMed
