

Furin targeted drug delivery for treatment of rhabdomyosarcoma in a mouse model
- PMID: 20454619
- PMCID: PMC2862740
- DOI: 10.1371/journal.pone.0010445
Furin targeted drug delivery for treatment of rhabdomyosarcoma in a mouse model
Abstract
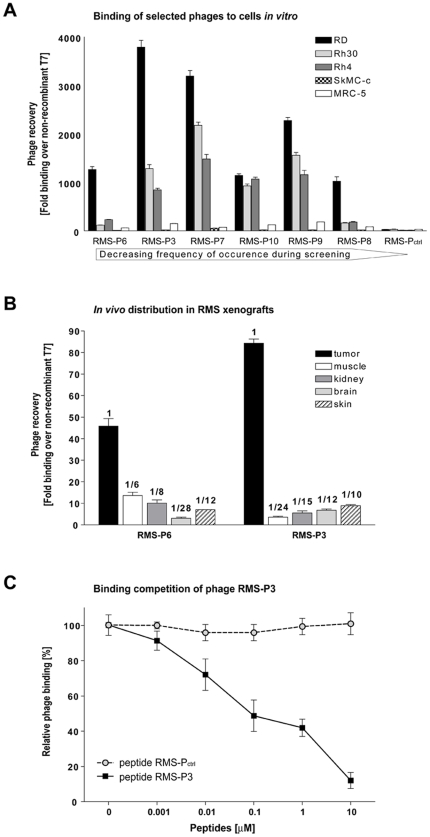
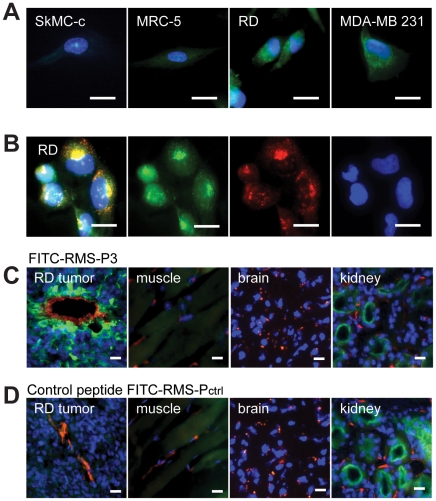
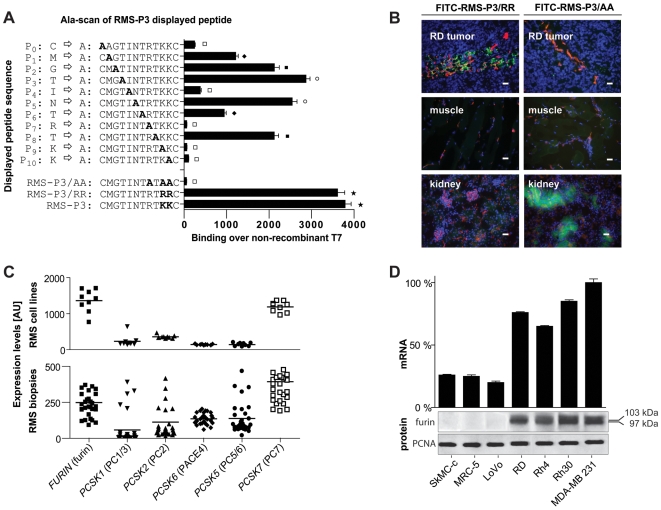
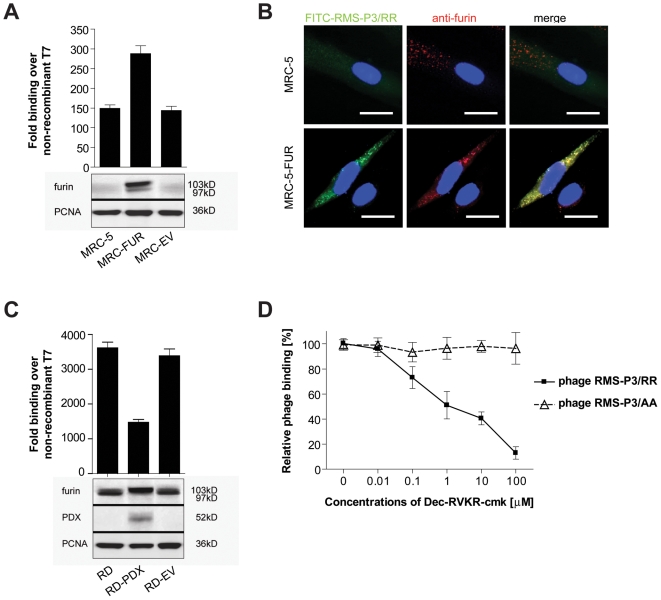
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children. Improvement of treatment efficacy and decreased side effects through tumor-targeted drug delivery would be desirable. By panning with a phage-displayed cyclic random peptide library we selected a peptide with strong affinity for RMS in vitro and in vivo. The peptide minimal binding motif Arg-X-(Arg/Lys)(Arg/Lys) identified by alanine-scan, suggested the target receptor to be a proprotein convertase (PC). Expression profiling of all PCs in RMS biopsies and cell lines revealed consistent high expression levels for the membrane-bound furin and PC7. Direct binding of RMS-P3 peptide to furin was demonstrated by affinity chromatography and supported by activity and colocalization studies. Treatment of RMS in mice with doxorubicin coupled to the targeting peptide resulted in a two-fold increase in therapeutic efficacy compared to doxorubicin treatment alone. Our findings indicate surface-furin binding as novel mechanism for therapeutic cell penetration which needs to be further investigated. Furthermore, this work demonstrates that specific targeting of membrane-bound furin in tumors is possible for and suggests that RMS and other tumors might benefit from proprotein convertases targeted drug delivery.
Conflict of interest statement
Figures
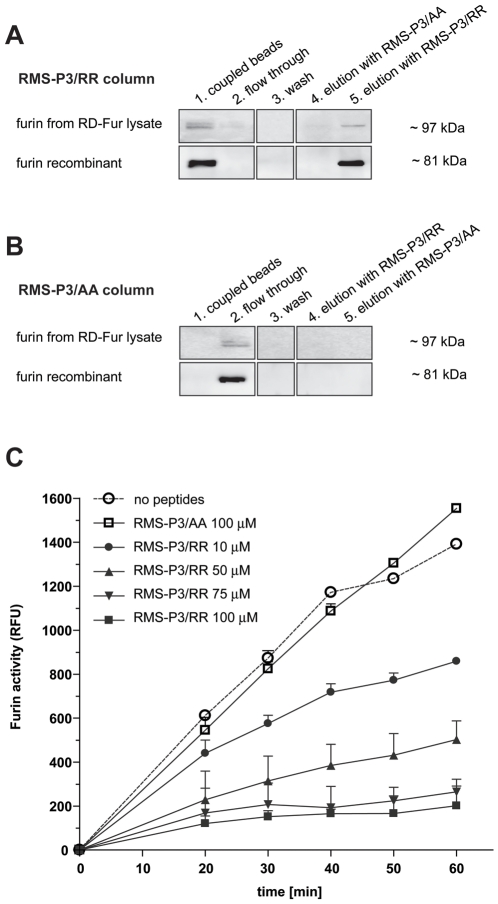
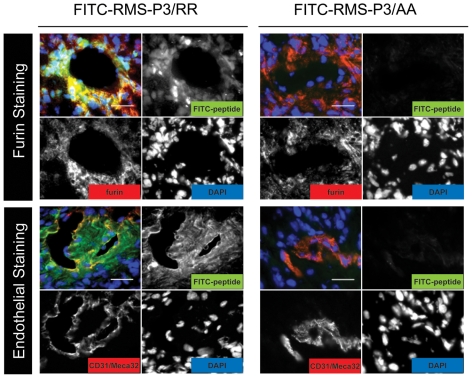
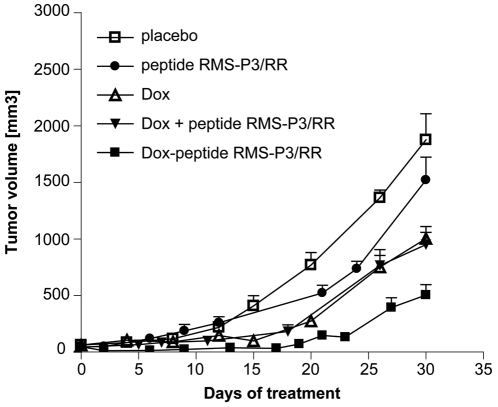
References
-
- De Giovanni C, Landuzzi L, Nicoletti G, Lollini PL, Nanni P. Molecular and cellular biology of rhabdomyosarcoma. Future Oncol. 2009;5:1449–1475. - PubMed
-
- Breitfeld PP, Meyer WH. Rhabdomyosarcoma: new windows of opportunity. Oncologist. 2005;10:518–527. - PubMed
-
- Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–1157. - PubMed
-
- Cortez-Retamozo V, Backmann N, Senter PD, Wernery U, De Baetselier P, et al. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004;64:2853–2857. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
