

Guidelines for identifying homologous recombination events in influenza A virus
- PMID: 20454662
- PMCID: PMC2862710
- DOI: 10.1371/journal.pone.0010434
Guidelines for identifying homologous recombination events in influenza A virus
Abstract
The rapid evolution of influenza viruses occurs both clonally and non-clonally through a variety of genetic mechanisms and selection pressures. The non-clonal evolution of influenza viruses comprises relatively frequent reassortment among gene segments and a more rarely reported process of non-homologous RNA recombination. Homologous RNA recombination within segments has been proposed as a third such mechanism, but to date the evidence for the existence of this process among influenza viruses has been both weak and controversial. As homologous recombination has not yet been demonstrated in the laboratory, supporting evidence, if it exists, may come primarily from patterns of phylogenetic incongruence observed in gene sequence data. Here, we review the necessary criteria related to laboratory procedures and sample handling, bioinformatic analysis, and the known ecology and evolution of influenza viruses that need to be met in order to confirm that a homologous recombination event occurred in the history of a set of sequences. To determine if these criteria have an effect on recombination analysis, we gathered 8307 publicly available full-length sequences of influenza A segments and divided them into those that were sequenced via the National Institutes of Health Influenza Genome Sequencing Project (IGSP) and those that were not. As sample handling and sequencing are executed to a very high standard in the IGSP, these sequences should be less likely to be exposed to contamination by other samples or by laboratory strains, and thus should not exhibit laboratory-generated signals of homologous recombination. Our analysis shows that the IGSP data set contains only two phylogenetically-supported single recombinant sequences and no recombinant clades. In marked contrast, the non-IGSP data show a very large amount of potential recombination. We conclude that the presence of false positive signals in the non-IGSP data is more likely than false negatives in the IGSP data, and that given the evidence to date, homologous recombination seems to play little or no role in the evolution of influenza A viruses.
Conflict of interest statement
Figures
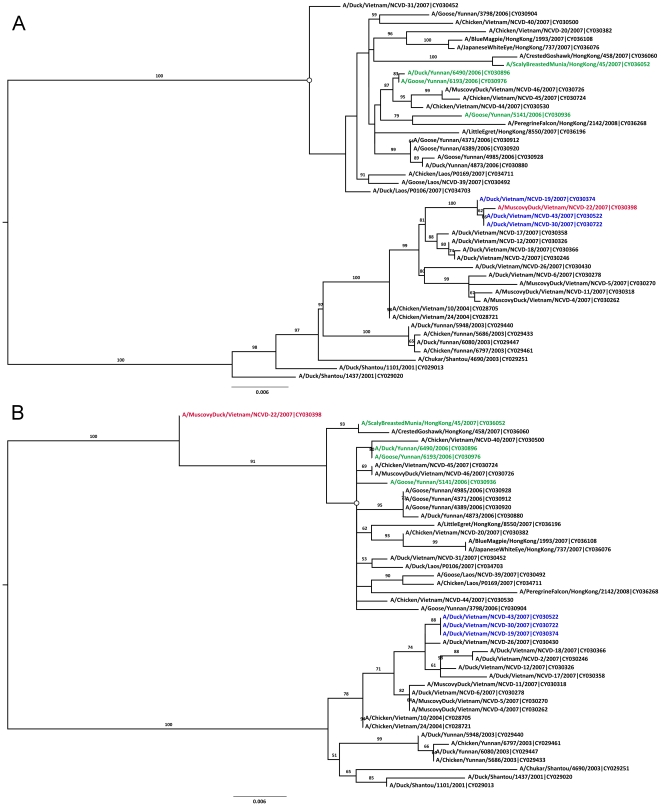
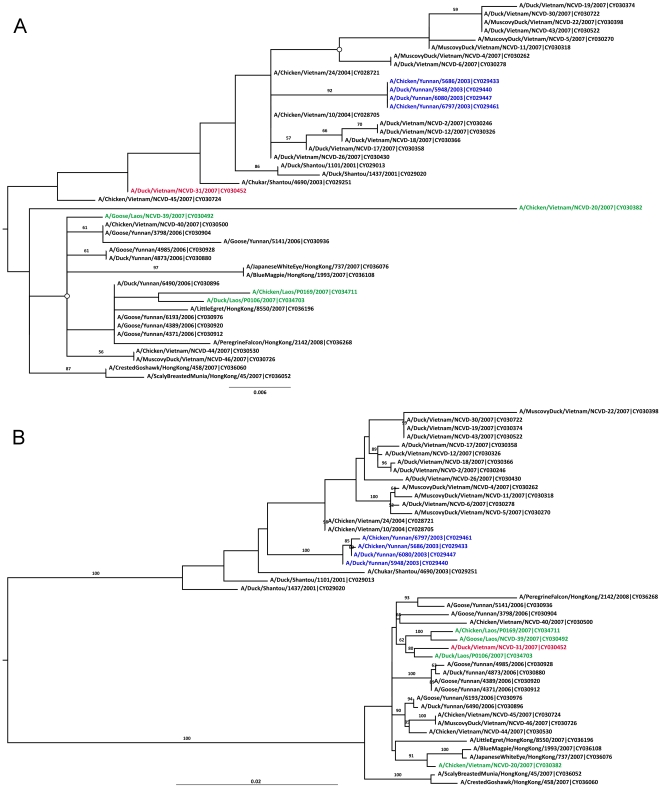
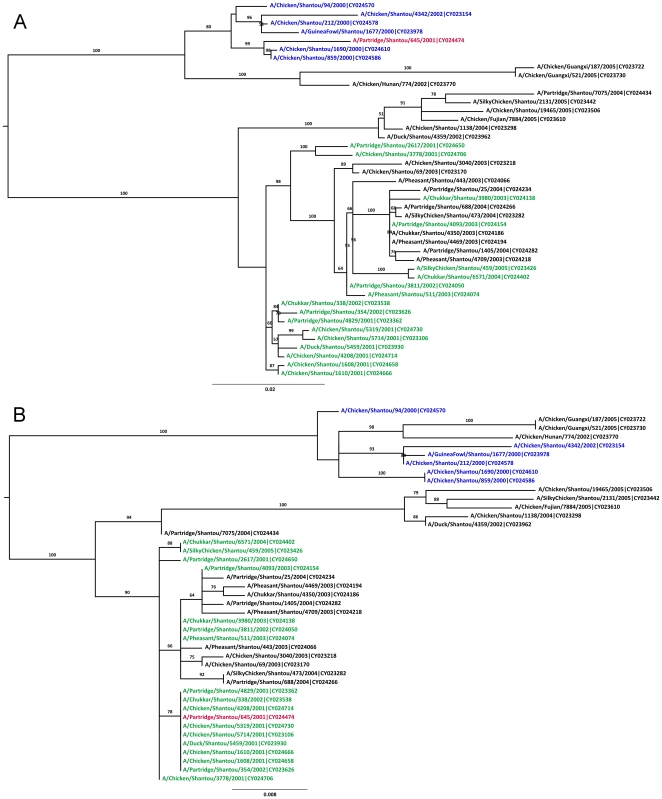
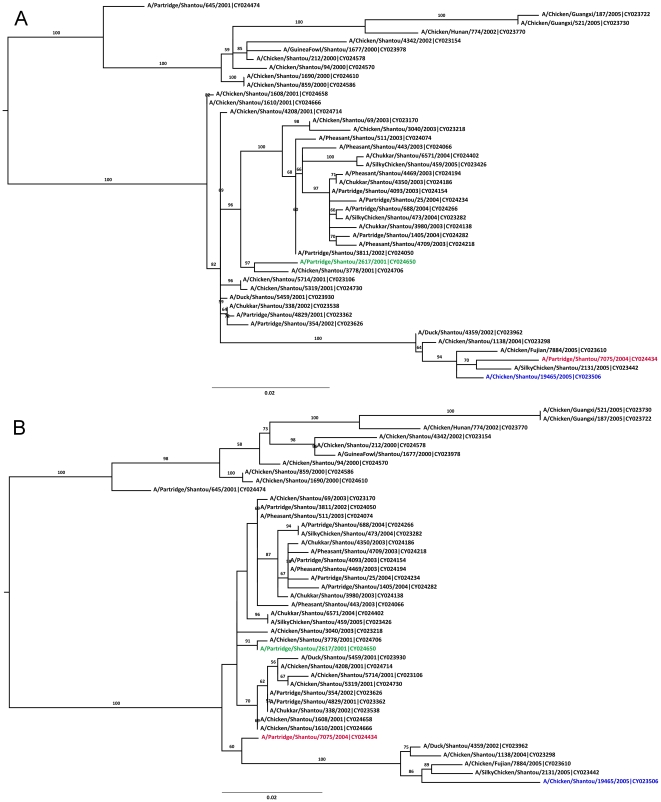
References
-
- Aoki FY, Boivin G, Roberts N. Influenza virus susceptibility and resistance to oseltamivir. Antiviral Therapy. 2007;12:603–616. - PubMed
-
- Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, et al. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis. 2007;196:249–257. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
