

Pro-autophagic signal induction by bacterial pore-forming toxins
- PMID: 20454906
- PMCID: PMC2955911
- DOI: 10.1007/s00430-010-0163-0
Pro-autophagic signal induction by bacterial pore-forming toxins
Abstract
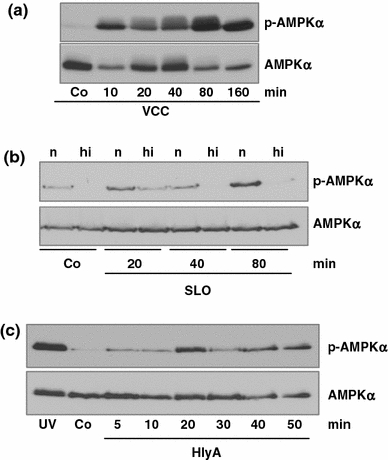
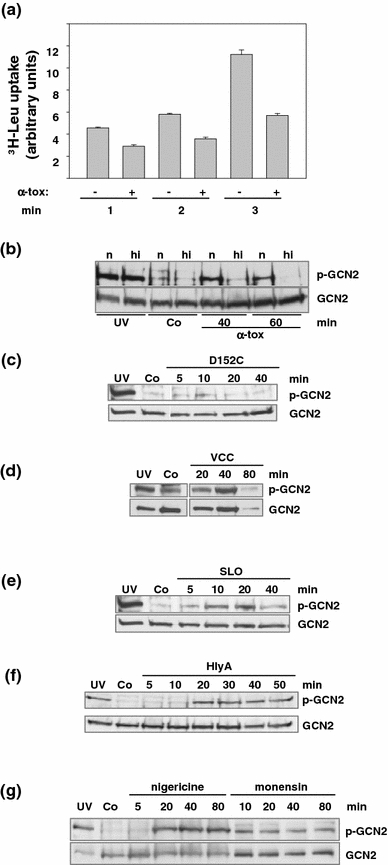
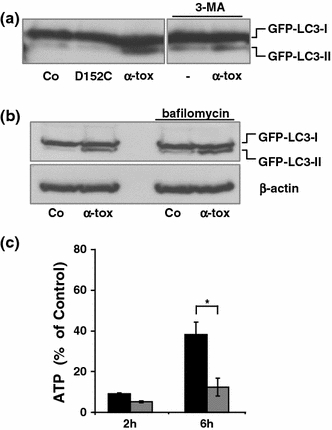
Pore-forming toxins (PFT) comprise a large, structurally heterogeneous group of bacterial protein toxins. Nucleated target cells mount complex responses which allow them to survive moderate membrane damage by PFT. Autophagy has recently been implicated in responses to various PFT, but how this process is triggered is not known, and the significance of the phenomenon is not understood. Here, we show that S. aureus α-toxin, Vibrio cholerae cytolysin, streptolysin O and E. coli haemolysin activate two pathways leading to autophagy. The first pathway is triggered via AMP-activated protein kinase (AMPK). AMPK is a major energy sensor which induces autophagy by inhibiting the target of rapamycin complex 1 (TORC1) in response to a drop of the cellular ATP/AMP-ratio, as is also observed in response to membrane perforation. The second pathway is activated by the conserved eIF2α-kinase GCN2, which causes global translational arrest and promotes autophagy in response to starvation. The latter could be accounted for by impaired amino acid transport into target cells. Notably, PKR, an eIF2α-kinase which has been implicated in autophagy induction during viral infection, was also activated upon membrane perforation, and evidence was obtained that phosphorylation of eIF2α is required for the accumulation of autophagosomes in α-toxin-treated cells. Treatment with 3-methyl-adenine inhibited autophagy and disrupted the ability of cells to recover from sublethal attack by S. aureus α-toxin. We propose that PFT induce pro-autophagic signals through membrane perforation-dependent nutrient and energy depletion, and that an important function of autophagy in this context is to maintain metabolic homoeostasis.
Figures





Similar articles
-
Modulation of translation and induction of autophagy by bacterial exoproducts.Med Microbiol Immunol. 2012 Nov;201(4):409-18. doi: 10.1007/s00430-012-0271-0. Epub 2012 Sep 19. Med Microbiol Immunol. 2012. PMID: 22991039 Free PMC article. Review.
-
DNA-dependent protein kinase regulates lysosomal AMP-dependent protein kinase activation and autophagy.Autophagy. 2020 Oct;16(10):1871-1888. doi: 10.1080/15548627.2019.1710430. Epub 2020 Jan 26. Autophagy. 2020. PMID: 31983282 Free PMC article.
-
AMPK Inhibits ULK1-Dependent Autophagosome Formation and Lysosomal Acidification via Distinct Mechanisms.Mol Cell Biol. 2018 Apr 30;38(10):e00023-18. doi: 10.1128/MCB.00023-18. Print 2018 May 15. Mol Cell Biol. 2018. PMID: 29507183 Free PMC article.
-
The GST-BHMT assay reveals a distinct mechanism underlying proteasome inhibition-induced macroautophagy in mammalian cells.Autophagy. 2015;11(5):812-32. doi: 10.1080/15548627.2015.1034402. Autophagy. 2015. PMID: 25984893 Free PMC article.
-
WNK1 is an unexpected autophagy inhibitor.Autophagy. 2017 May 4;13(5):969-970. doi: 10.1080/15548627.2017.1286431. Epub 2017 Feb 15. Autophagy. 2017. PMID: 28282258 Free PMC article. Review.
Cited by
-
Translation inhibition and metabolic stress pathways in the host response to bacterial pathogens.Nat Rev Microbiol. 2013 Jun;11(6):365-9. doi: 10.1038/nrmicro3029. Epub 2013 May 13. Nat Rev Microbiol. 2013. PMID: 23669888 Review.
-
Listeriolysin O: from bazooka to Swiss army knife.Philos Trans R Soc Lond B Biol Sci. 2017 Aug 5;372(1726):20160222. doi: 10.1098/rstb.2016.0222. Philos Trans R Soc Lond B Biol Sci. 2017. PMID: 28630160 Free PMC article. Review.
-
The bacterial and cellular determinants controlling the recruitment of mTOR to the Salmonella-containing vacuole.Biol Open. 2012 Dec 15;1(12):1215-25. doi: 10.1242/bio.20122840. Epub 2012 Oct 4. Biol Open. 2012. PMID: 23259056 Free PMC article.
-
Phobalysin: Fisheye View of Membrane Perforation, Repair, Chemotaxis and Adhesion.Toxins (Basel). 2019 Jul 16;11(7):412. doi: 10.3390/toxins11070412. Toxins (Basel). 2019. PMID: 31315179 Free PMC article. Review.
-
GCN2 controls the cellular checkpoint: potential target for regulating inflammation.Cell Death Discov. 2018 Feb 14;4:20. doi: 10.1038/s41420-017-0022-5. eCollection 2018 Dec. Cell Death Discov. 2018. PMID: 29531817 Free PMC article. No abstract available.
References
-
- Bhakdi S, Tranum-Jensen J. Membrane damage by channel-forming proteins: staphylococcal alpha-toxin, streptolysin-O and the C5b–9 complement complex. Biochem Soc Symp. 1985;50:221–233. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
