

Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome
- PMID: 20457915
- PMCID: PMC2906866
- DOI: 10.1073/pnas.0914118107
Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome
Abstract
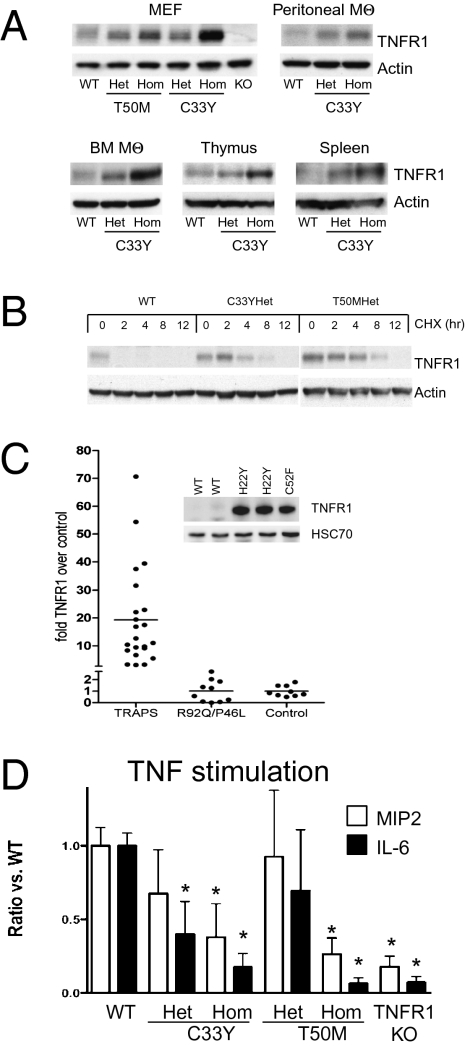
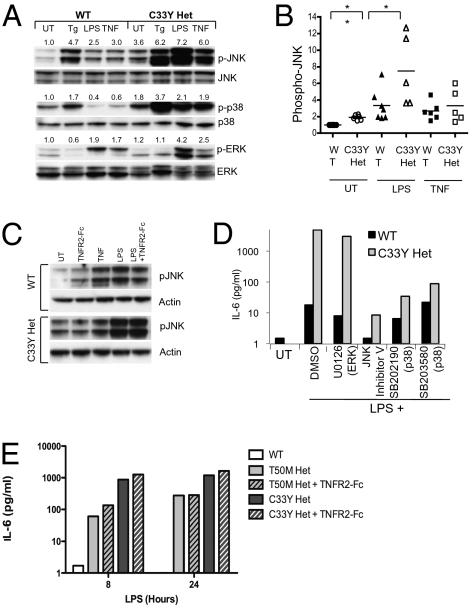
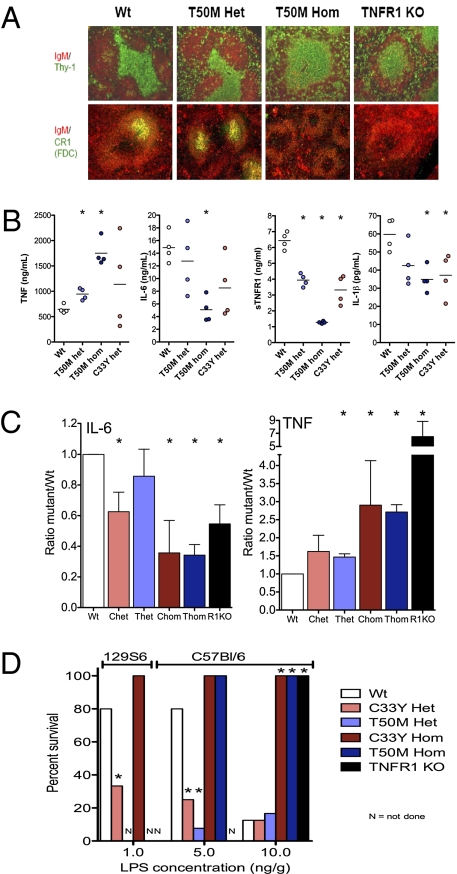
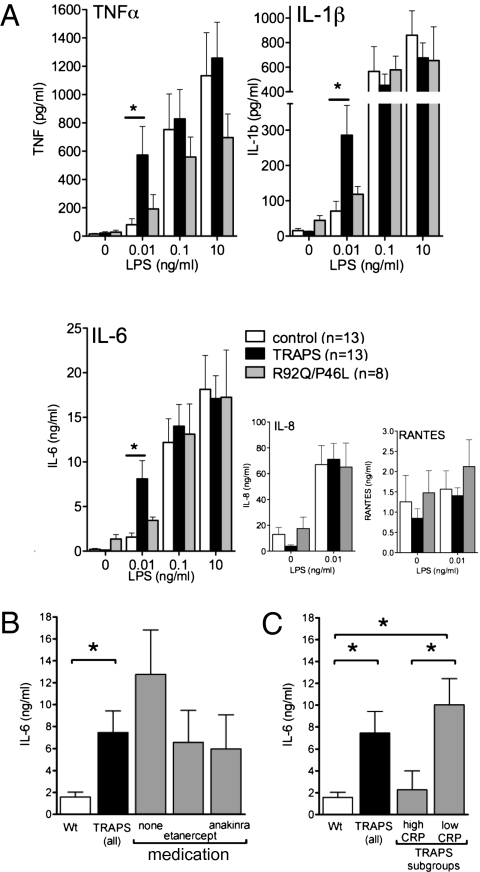
TNF, acting through p55 tumor necrosis factor receptor 1 (TNFR1), contributes to the pathogenesis of many inflammatory diseases. TNFR-associated periodic syndrome (TRAPS, OMIM 142680) is an autosomal dominant autoinflammatory disorder characterized by prolonged attacks of fevers, peritonitis, and soft tissue inflammation. TRAPS is caused by missense mutations in the extracellular domain of TNFR1 that affect receptor folding and trafficking. These mutations lead to loss of normal function rather than gain of function, and thus the pathogenesis of TRAPS is an enigma. Here we show that mutant TNFR1 accumulates intracellularly in peripheral blood mononuclear cells of TRAPS patients and in multiple cell types from two independent lines of knockin mice harboring TRAPS-associated TNFR1 mutations. Mutant TNFR1 did not function as a surface receptor for TNF but rather enhanced activation of MAPKs and secretion of proinflammatory cytokines upon stimulation with LPS. Enhanced inflammation depended on autocrine TNF secretion and WT TNFR1 in mouse and human myeloid cells but not in fibroblasts. Heterozygous TNFR1-mutant mice were hypersensitive to LPS-induced septic shock, whereas homozygous TNFR1-mutant mice resembled TNFR1-deficient mice and were resistant to septic shock. Thus WT and mutant TNFR1 act in concert from distinct cellular locations to potentiate inflammation in TRAPS. These findings establish a mechanism of pathogenesis in autosomal dominant diseases where full expression of the disease phenotype depends on functional cooperation between WT and mutant proteins and also may explain partial responses of TRAPS patients to TNF blockade.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Inflammation: New insights into the pathogenesis of TRAPS.Nat Rev Rheumatol. 2010 Jul;6(7):384. doi: 10.1038/nrrheum.2010.95. Nat Rev Rheumatol. 2010. PMID: 20614507 No abstract available.
References
-
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. - PubMed
-
- Chen G, Goeddel DV. TNF-R1 signaling: A beautiful pathway. Science. 2002;296:1634–1635. - PubMed
-
- McDermott MF, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–144. - PubMed
-
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
