

Most transcription factor binding sites are in a few mosaic classes of the human genome
- PMID: 20459624
- PMCID: PMC2881025
- DOI: 10.1186/1471-2164-11-286
Most transcription factor binding sites are in a few mosaic classes of the human genome
Abstract
Background: Many algorithms for finding transcription factor binding sites have concentrated on the characterisation of the binding site itself: and these algorithms lead to a large number of false positive sites. The DNA sequence which does not bind has been modeled only to the extent necessary to complement this formulation.
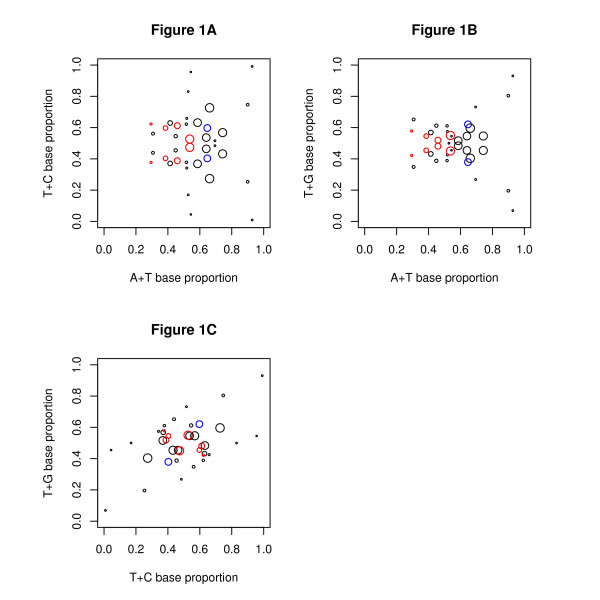
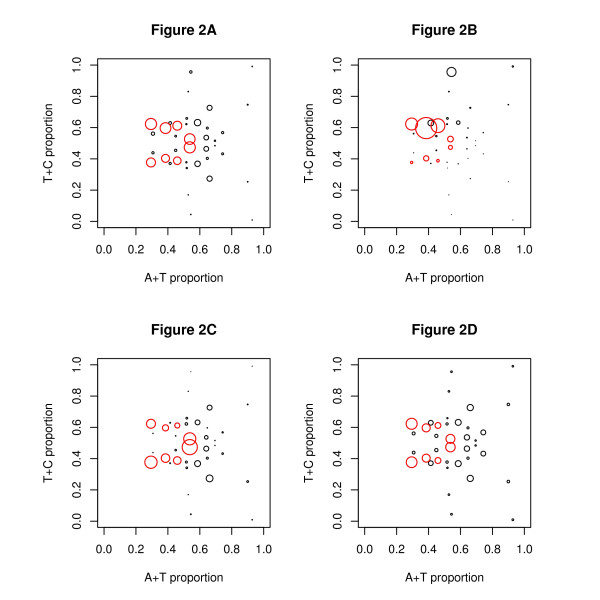
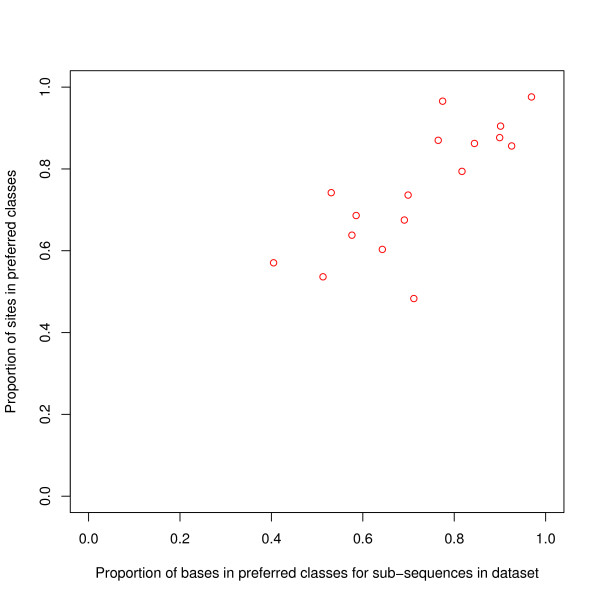
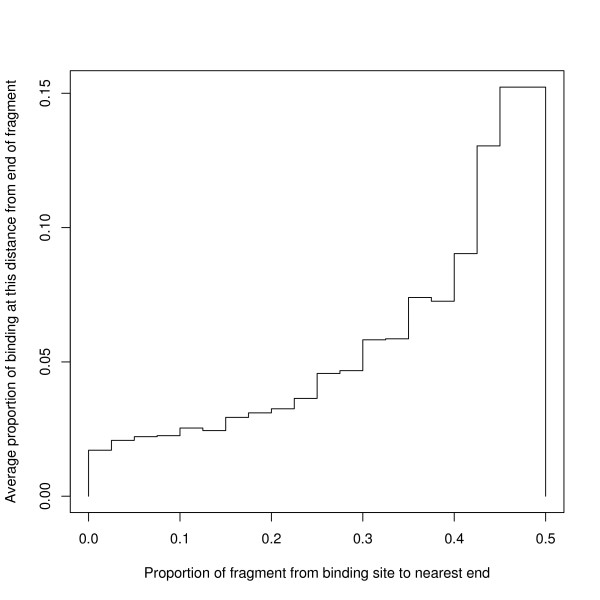
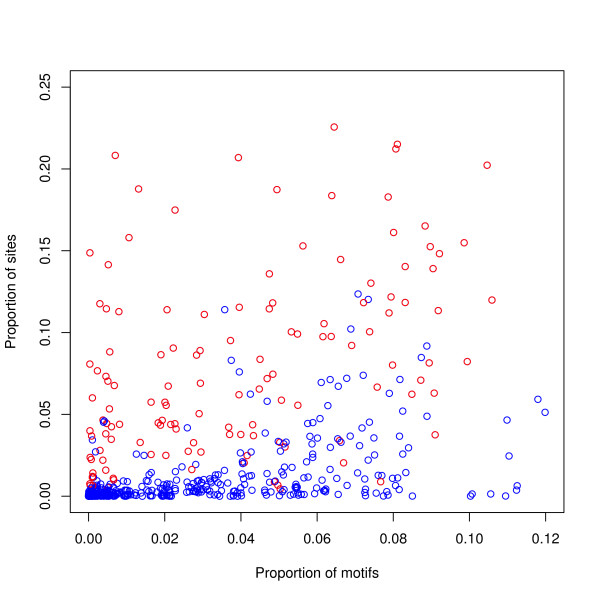
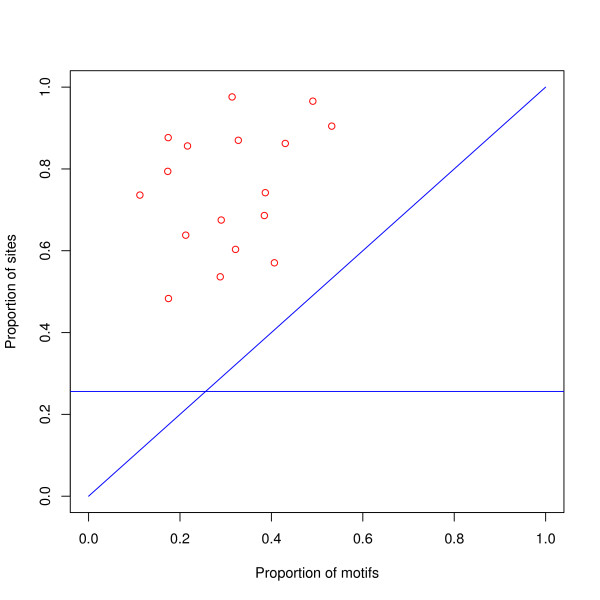
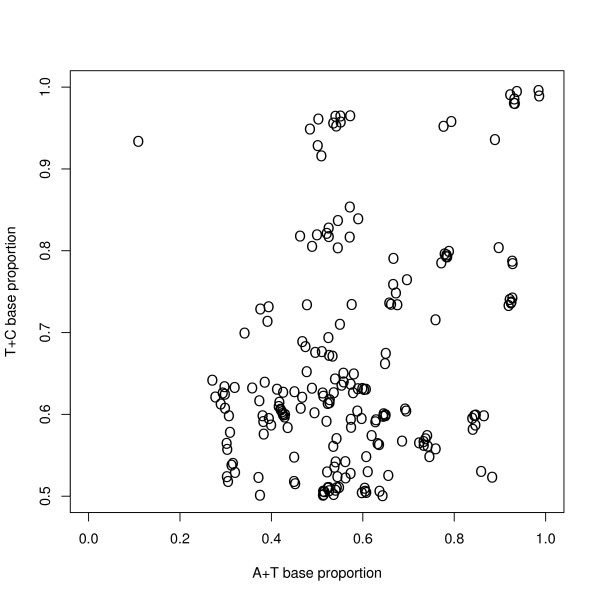
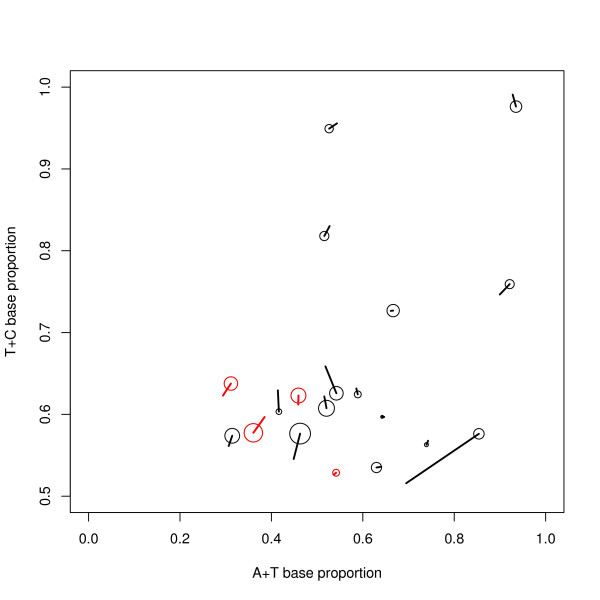
Results: We find that the human genome may be described by 19 pairs of mosaic classes, each defined by its base frequencies, (or more precisely by the frequencies of doublets), so that typically a run of 10 to 100 bases belongs to the same class. Most experimentally verified binding sites are in the same four pairs of classes. In our sample of seventeen transcription factors - taken from different families of transcription factors - the average proportion of sites in this subset of classes was 75%, with values for individual factors ranging from 48% to 98%. By contrast these same classes contain only 26% of the bases of the genome and only 31% of occurrences of the motifs of these factors - that is places where one might expect the factors to bind. These results are not a consequence of the class composition in promoter regions.
Conclusions: This method of analysis will help to find transcription factor binding sites and assist with the problem of false positives. These results also imply a profound difference between the mosaic classes.
Figures
Similar articles
-
Ab initio identification of putative human transcription factor binding sites by comparative genomics.BMC Bioinformatics. 2005 May 2;6:110. doi: 10.1186/1471-2105-6-110. BMC Bioinformatics. 2005. PMID: 15865625 Free PMC article.
-
Two different classes of co-occurring motif pairs found by a novel visualization method in human promoter regions.BMC Genomics. 2008 Mar 1;9:112. doi: 10.1186/1471-2164-9-112. BMC Genomics. 2008. PMID: 18312685 Free PMC article.
-
Position specific variation in the rate of evolution in transcription factor binding sites.BMC Evol Biol. 2003 Aug 28;3:19. doi: 10.1186/1471-2148-3-19. Epub 2003 Aug 28. BMC Evol Biol. 2003. PMID: 12946282 Free PMC article.
-
Integrating genomic data to predict transcription factor binding.Genome Inform. 2005;16(1):83-94. Genome Inform. 2005. PMID: 16362910
-
An overview of the computational analyses and discovery of transcription factor binding sites.Methods Mol Biol. 2010;674:1-22. doi: 10.1007/978-1-60761-854-6_1. Methods Mol Biol. 2010. PMID: 20827582 Review.
Cited by
-
Validation of skeletal muscle cis-regulatory module predictions reveals nucleotide composition bias in functional enhancers.PLoS Comput Biol. 2011 Dec;7(12):e1002256. doi: 10.1371/journal.pcbi.1002256. Epub 2011 Dec 1. PLoS Comput Biol. 2011. PMID: 22144875 Free PMC article.
References
-
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, Wheeler R, Wong B, Drenkow J, Yamanaka M, Patel S, Brubaker S, Tammana H, Helt G, Struhl K, Gingeras TR. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116(4):499–509. doi: 10.1016/S0092-8674(04)00127-8. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
