

Highly-multiplexed barcode sequencing: an efficient method for parallel analysis of pooled samples
- PMID: 20460461
- PMCID: PMC2910071
- DOI: 10.1093/nar/gkq368
Highly-multiplexed barcode sequencing: an efficient method for parallel analysis of pooled samples
Abstract
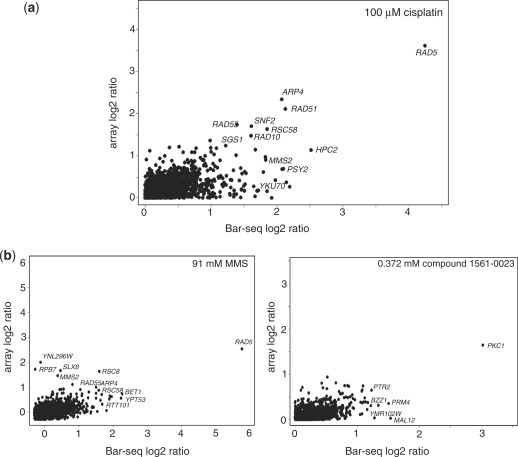
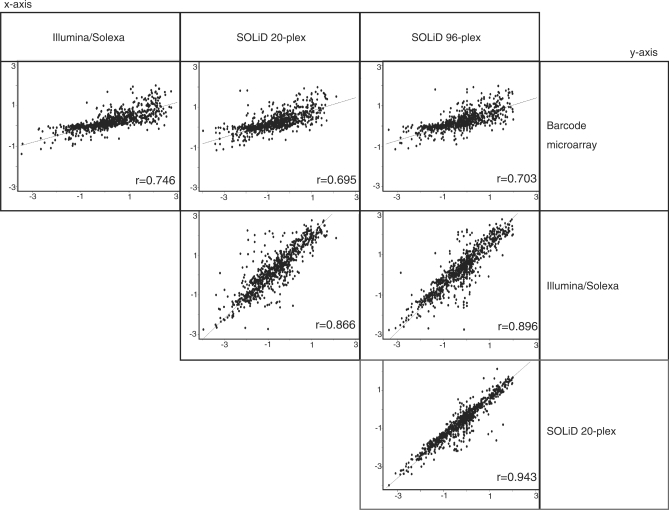
Next-generation sequencing has proven an extremely effective technology for molecular counting applications where the number of sequence reads provides a digital readout for RNA-seq, ChIP-seq, Tn-seq and other applications. The extremely large number of sequence reads that can be obtained per run permits the analysis of increasingly complex samples. For lower complexity samples, however, a point of diminishing returns is reached when the number of counts per sequence results in oversampling with no increase in data quality. A solution to making next-generation sequencing as efficient and affordable as possible involves assaying multiple samples in a single run. Here, we report the successful 96-plexing of complex pools of DNA barcoded yeast mutants and show that such 'Bar-seq' assessment of these samples is comparable with data provided by barcode microarrays, the current benchmark for this application. The cost reduction and increased throughput permitted by highly multiplexed sequencing will greatly expand the scope of chemogenomics assays and, equally importantly, the approach is suitable for other sequence counting applications that could benefit from massive parallelization.
Figures
References
-
- Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, Fox P, Glasscock JI, Hickenbotham M, Huang W, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat. Methods. 2008;5:183–188. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
