

Metabolomic and transcriptomic stress response of Escherichia coli
- PMID: 20461071
- PMCID: PMC2890322
- DOI: 10.1038/msb.2010.18
Metabolomic and transcriptomic stress response of Escherichia coli
Abstract
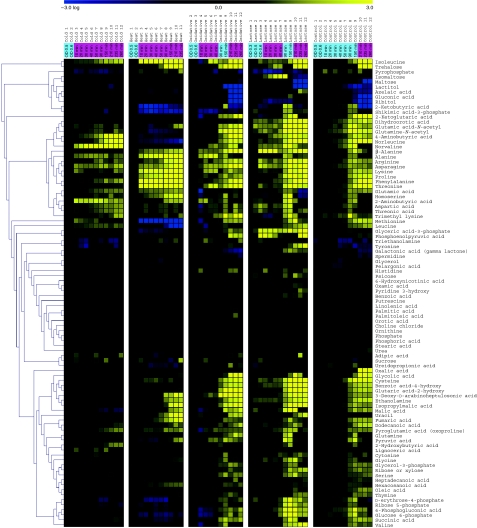
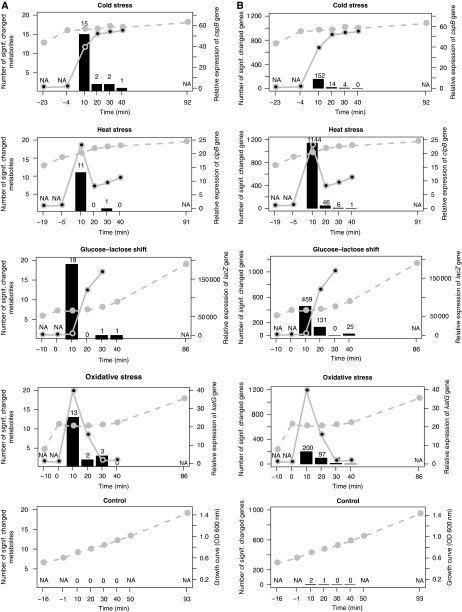
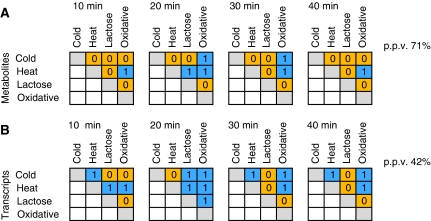
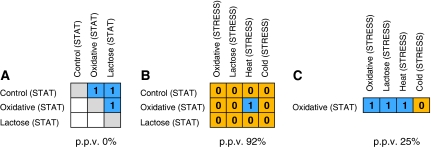
Environmental fluctuations lead to a rapid adjustment of the physiology of Escherichia coli, necessitating changes on every level of the underlying cellular and molecular network. Thus far, the majority of global analyses of E. coli stress responses have been limited to just one level, gene expression. Here, we incorporate the metabolite composition together with gene expression data to provide a more comprehensive insight on system level stress adjustments by describing detailed time-resolved E. coli response to five different perturbations (cold, heat, oxidative stress, lactose diauxie, and stationary phase). The metabolite response is more specific as compared with the general response observed on the transcript level and is reflected by much higher specificity during the early stress adaptation phase and when comparing the stationary phase response to other perturbations. Despite these differences, the response on both levels still follows the same dynamics and general strategy of energy conservation as reflected by rapid decrease of central carbon metabolism intermediates coinciding with downregulation of genes related to cell growth. Application of co-clustering and canonical correlation analysis on combined metabolite and transcript data identified a number of significant condition-dependent associations between metabolites and transcripts. The results confirm and extend existing models about co-regulation between gene expression and metabolites demonstrating the power of integrated systems oriented analysis.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
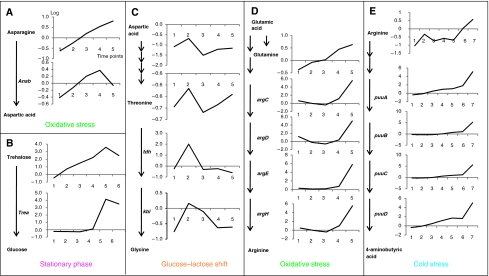
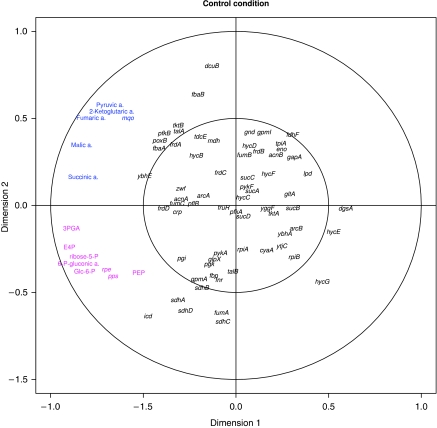
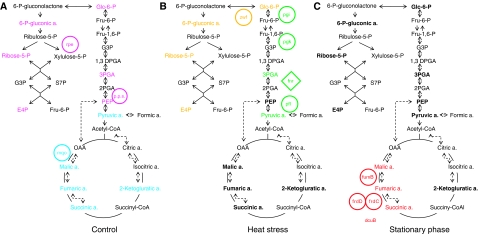
References
-
- Agresti A (2002) Categorical Data Analysis. New York: John Wiley & Sons
-
- Barker MM, Gaal T, Gourse RL (2001) Mechanism of regulation of transcription initiation by ppGpp. II. Models for positive control based on properties of RNAP mutants and competition for rnap. J Mol Biol 305: 689–702 - PubMed
-
- Berg JM, Tymoczko JL, Stryer L (2006) Biochemistry. New York: W. H. Freeman
-
- Bolten CJ, Kiefer P, Letisse F, Portais JC, Wittmann C (2007) Sampling for metabolome analysis of microorganisms. Anal Chem 79: 3843–3849 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
