

High frequency of COH1 intragenic deletions and duplications detected by MLPA in patients with Cohen syndrome
- PMID: 20461111
- PMCID: PMC2987453
- DOI: 10.1038/ejhg.2010.59
High frequency of COH1 intragenic deletions and duplications detected by MLPA in patients with Cohen syndrome
Abstract
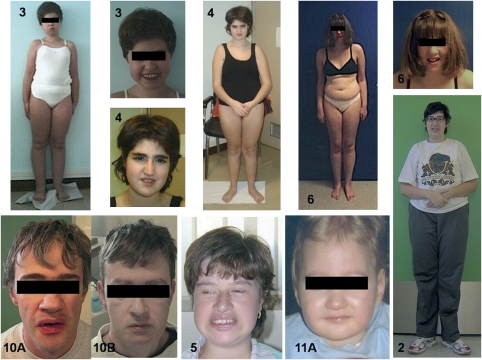
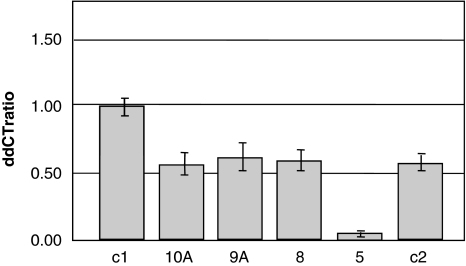
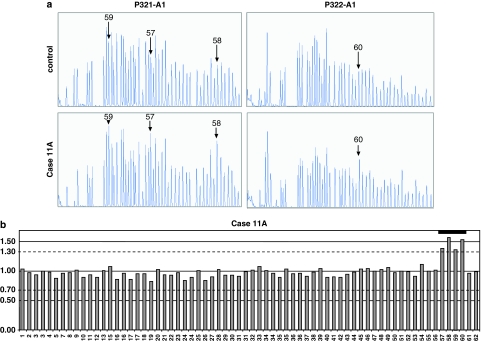
Cohen syndrome is a rare, clinically variable autosomal recessive disorder characterized by mental retardation, postnatal microcephaly, facial dysmorphisms, ocular abnormalities and intermittent neutropenia. Mutations in the COH1 gene have been found in patients from different ethnic origins. However, a high percentage of patients have only one or no mutated allele. To investigate whether COH1 copy number changes account for missed mutations, we used multiplex ligation-dependent probe amplification (MLPA) to test a group of 14 patients with Cohen syndrome. This analysis has allowed us to identify multi-exonic deletions in 11 alleles and duplications in 4 alleles. Considering our previous study, COH1 copy number variations represent 42% of total mutated alleles. To our knowledge, COH1 intragenic duplications have never been reported in Cohen syndrome. The three duplications encompassed exons 4-13, 20-30 and 57-60, respectively. Interestingly, four deletions showed the same exon coverage (exons 6-16) with respect to a deletion recently reported in a large Greek consanguineous family. Haplotype analysis suggested a possible founder effect in the Mediterranean basin. The use of MLPA was therefore crucial in identifying mutated alleles undetected by traditional techniques and in defining the extent of the deletions/duplications. Given the high percentage of identified copy number variations, we suggest that this technique could be used as the initial screening method for molecular diagnosis of Cohen syndrome.
Figures


References
-
- Cohen MM, Jr, Hall BD, Smith DW, Graham CB, Lampert KJ. A new syndrome with hypotonia, obesity, mental deficiency, and facial, oral, ocular, and limb anomalies. J Pediatr. 1973;83:280–284. - PubMed
-
- Carey JC, Hall BD. Confirmation of the Cohen syndrome. J Pediatr. 1978;93:239–244. - PubMed
-
- Norio R, Raitta C, Lindahl E. Further delineation of the Cohen syndrome; report on chorioretinal dystrophy, leukopenia and consanguinity. Clin Genet. 1984;25:1–14. - PubMed
-
- Velayos-Baeza A, Vettori A, Copley RR, Dobson-Stone C, Monaco AP. Analysis of the human VPS13 gene family. Genomics. 2004;84:536–549. - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
