

TumorBoost: normalization of allele-specific tumor copy numbers from a single pair of tumor-normal genotyping microarrays
- PMID: 20462408
- PMCID: PMC2894037
- DOI: 10.1186/1471-2105-11-245
TumorBoost: normalization of allele-specific tumor copy numbers from a single pair of tumor-normal genotyping microarrays
Abstract
Background: High-throughput genotyping microarrays assess both total DNA copy number and allelic composition, which makes them a tool of choice for copy number studies in cancer, including total copy number and loss of heterozygosity (LOH) analyses. Even after state of the art preprocessing methods, allelic signal estimates from genotyping arrays still suffer from systematic effects that make them difficult to use effectively for such downstream analyses.
Results: We propose a method, TumorBoost, for normalizing allelic estimates of one tumor sample based on estimates from a single matched normal. The method applies to any paired tumor-normal estimates from any microarray-based technology, combined with any preprocessing method. We demonstrate that it increases the signal-to-noise ratio of allelic signals, making it significantly easier to detect allelic imbalances.
Conclusions: TumorBoost increases the power to detect somatic copy-number events (including copy-neutral LOH) in the tumor from allelic signals of Affymetrix or Illumina origin. We also conclude that high-precision allelic estimates can be obtained from a single pair of tumor-normal hybridizations, if TumorBoost is combined with single-array preprocessing methods such as (allele-specific) CRMA v2 for Affymetrix or BeadStudio's (proprietary) XY-normalization method for Illumina. A bounded-memory implementation is available in the open-source and cross-platform R package aroma.cn, which is part of the Aroma Project (http://www.aroma-project.org/).
Figures
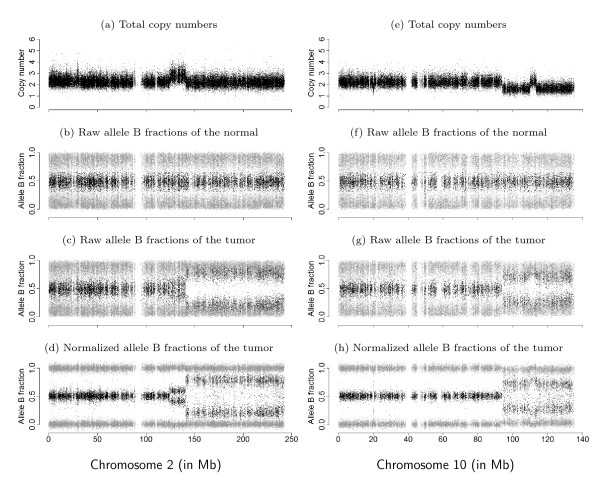
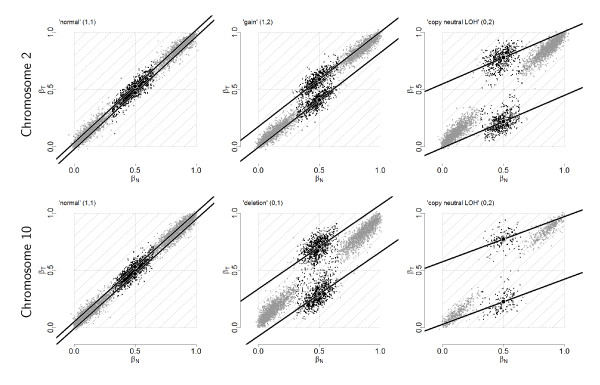
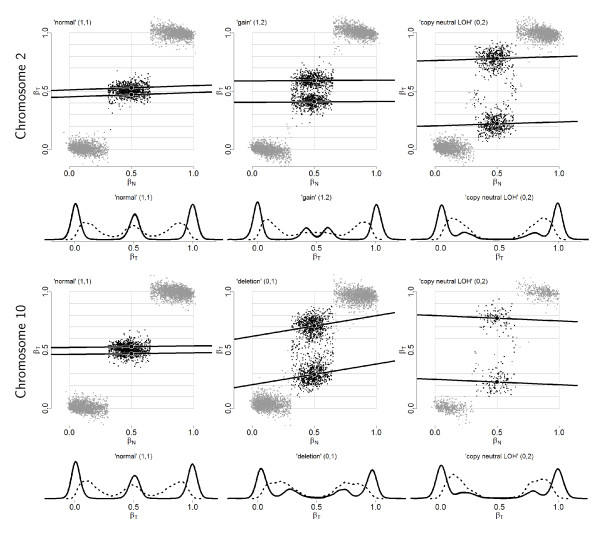
 ), and empirical densities of the raw (βT; dashed) and the normalized (; solid) allele B fractions for sample TCGA-23-1027. The same regions, SNPs and annotation as in Figure 2 are used.
), and empirical densities of the raw (βT; dashed) and the normalized (; solid) allele B fractions for sample TCGA-23-1027. The same regions, SNPs and annotation as in Figure 2 are used.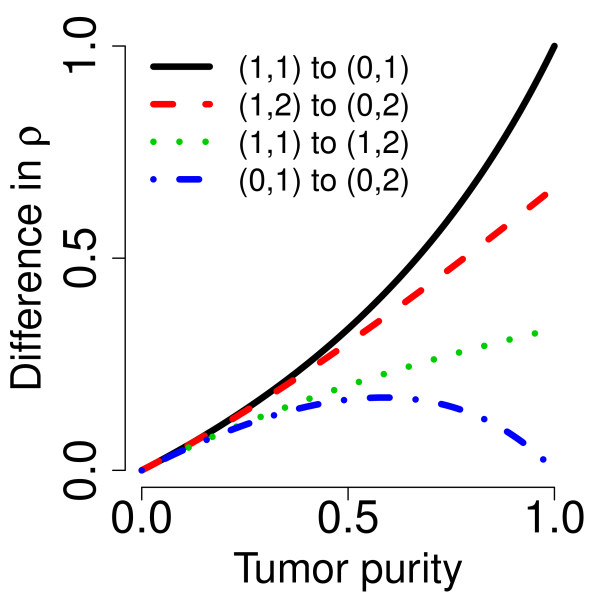
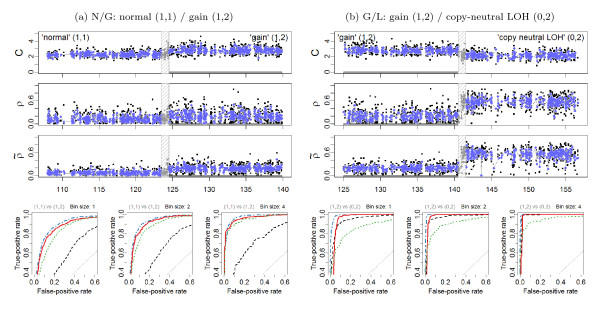
 ) heterozygous DHs, respectively. A 1000 kb safety region (dashed gray frame) around the change point is excluded from the evaluation. The full resolution data points are colored black and the binned (H = 4) ones are colored blue. The three panels in the bottom row show the ROC performance of the TCNs (dotted green) and the raw (dashed black) and normalized (solid red and dot-dashed blue for naive and population-based genotypes, respectively) DHs at the full resolution (H = 1; no binning), and after binning in non-overlapping windows of size H = 2 and H = 4 SNPs, respectively.
) heterozygous DHs, respectively. A 1000 kb safety region (dashed gray frame) around the change point is excluded from the evaluation. The full resolution data points are colored black and the binned (H = 4) ones are colored blue. The three panels in the bottom row show the ROC performance of the TCNs (dotted green) and the raw (dashed black) and normalized (solid red and dot-dashed blue for naive and population-based genotypes, respectively) DHs at the full resolution (H = 1; no binning), and after binning in non-overlapping windows of size H = 2 and H = 4 SNPs, respectively.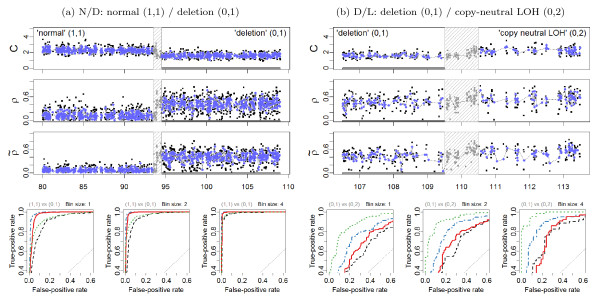
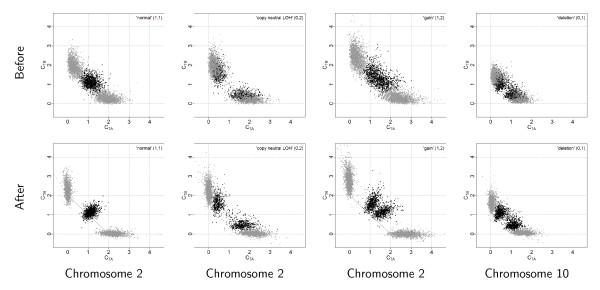
References
-
- Affymetrix Inc. Genome-Wide Human SNP Nsp/Sty 6.0 user guide. Affymetrix Inc; 2007. [Rev 1.]
-
- Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J, Cheung SW, Shen RM, Barker DL, Gunderson KL. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16(9):1136–1148. doi: 10.1101/gr.5402306. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
