

A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate
- PMID: 20462955
- PMCID: PMC2893996
- DOI: 10.1091/mbc.e10-01-0028
A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate
Abstract
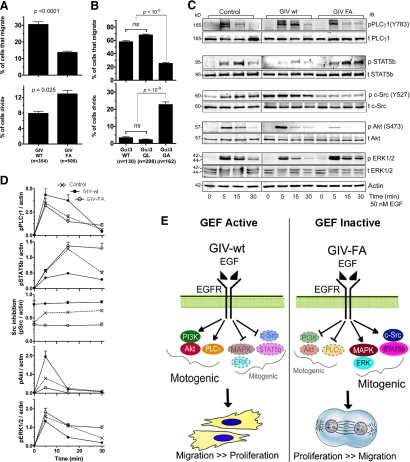
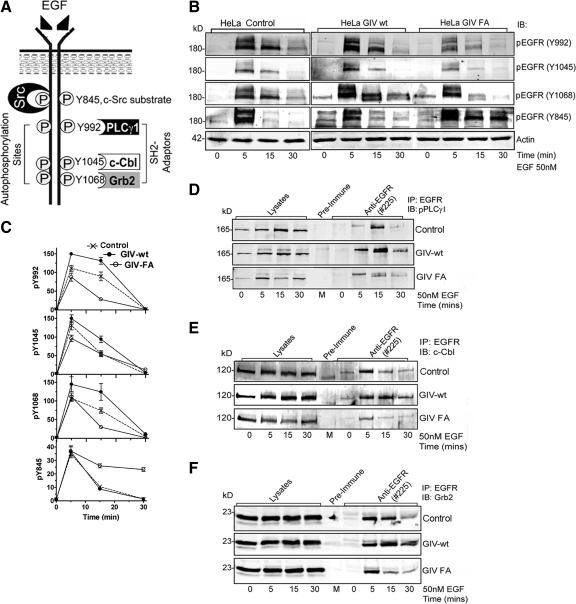
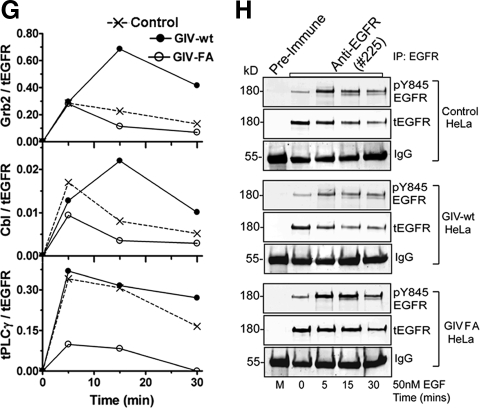
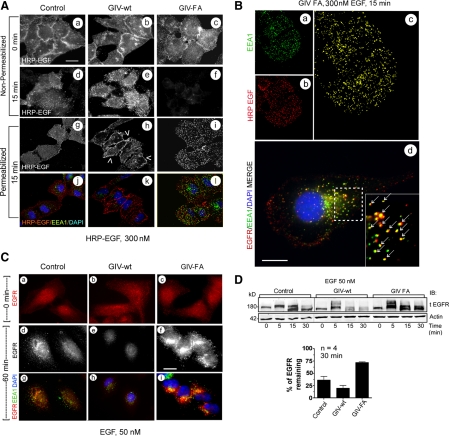
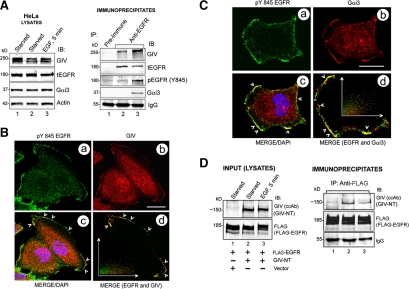
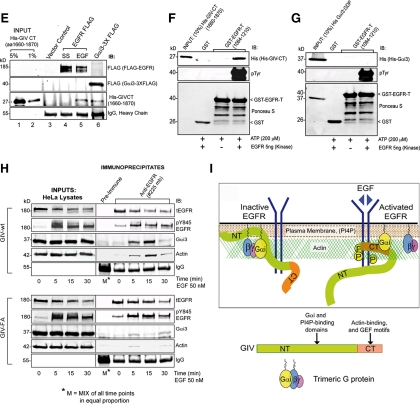
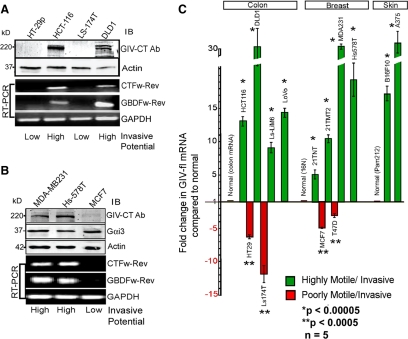
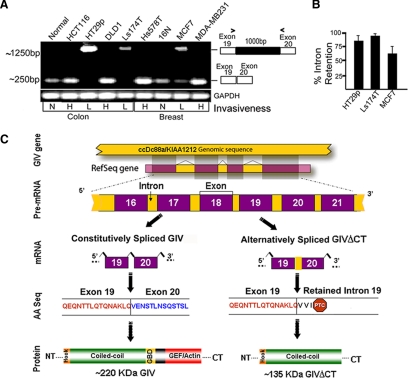
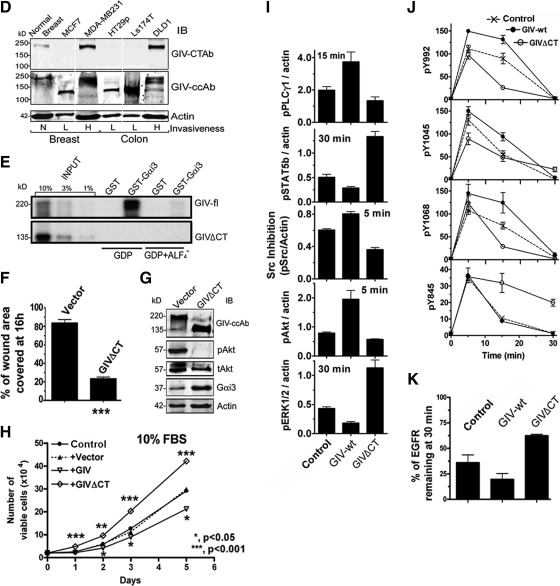
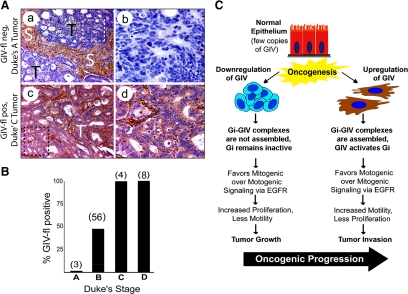
Cells respond to growth factors by either migrating or proliferating, but not both at the same time, a phenomenon termed migration-proliferation dichotomy. The underlying mechanism of this phenomenon has remained unknown. We demonstrate here that Galpha(i) protein and GIV, its nonreceptor guanine nucleotide exchange factor (GEF), program EGF receptor (EGFR) signaling and orchestrate this dichotomy. GIV directly interacts with EGFR, and when its GEF function is intact, a Galpha(i)-GIV-EGFR signaling complex assembles, EGFR autophosphorylation is enhanced, and the receptor's association with the plasma membrane (PM) is prolonged. Accordingly, PM-based motogenic signals (PI3-kinase-Akt and PLCgamma1) are amplified, and cell migration is triggered. In cells expressing a GEF-deficient mutant, the Galphai-GIV-EGFR signaling complex is not assembled, EGFR autophosphorylation is reduced, the receptor's association with endosomes is prolonged, mitogenic signals (ERK 1/2, Src, and STAT5) are amplified, and cell proliferation is triggered. In rapidly growing, poorly motile breast and colon cancer cells and in noninvasive colorectal carcinomas in situ in which EGFR signaling favors mitosis over motility, a GEF-deficient splice variant of GIV was identified. In slow growing, highly motile cancer cells and late invasive carcinomas, GIV is highly expressed and has an intact GEF motif. Thus, inclusion or exclusion of GIV's GEF motif, which activates Galphai, modulates EGFR signaling, generates migration-proliferation dichotomy, and most likely influences cancer progression.
Figures
References
-
- Anai M., et al. A novel protein kinase B (PKB)/AKT-binding protein enhances PKB kinase activity and regulates DNA synthesis. J. Biol. Chem. 2005;280:18525–18535. - PubMed
-
- Athale C., Mansury Y., Deisboeck T. S. Simulating the impact of a molecular ‘decision-process’ on cellular phenotype and multicellular patterns in brain tumors. J. Theor. Biol. 2005;233:469–481. - PubMed
-
- Ausprunk D. H., Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 1977;14:53–65. - PubMed
-
- Bagrodia S., Chackalaparampil I., Kmiecik T. E., Shalloway D. Altered tyrosine 527 phosphorylation and mitotic activation of p60c-src. Nature. 1991;349:172–175. - PubMed
-
- Band V., Zajchowski D., Swisshelm K., Trask D., Kulesa V., Cohen C., Connolly J., Sager R. Tumor progression in four mammary epithelial cell lines derived from the same patient. Cancer Res. 1990;50:7351–7357. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
